14 Genómica funcional de procariontes
El objetivo de esta sección es hacer un análisis de expresión génica bajo diferentes condiciones (expresión diferencial). Para efectos de este curso, la primera parte de esta sección explica cómo, a través de un experimento de RNA-Seq y el uso de programas bioinformáticos, somos capaces de generar la tabla de cuentas de la expresión transcripcional de cada gen en cada condición del experimento. La que usaremos para hacer los análisis estadísticos y detectar qué genes se expresan diferencialmente de forma significativa en una condición vs. otra y visualización de resultados en el software R.
14.1 Set de datos
Los datos usados en este flujo de trabajo corresponden a la secuenciación de genoma completo (WGS: whole genome sequencing) y transcriptoma (RNA-Seq) de Salmonella enterica subsp. enterica serovar Typhimurium, provenientes de los siguientes proyectos: BioProject Accession: PRJNA280335 y BioProject Accession: PRJNA357075, respectivamente.
BioProject es una base de datos de NCBI que almacena todos los datos biológicos relacionados a un proyecto o estudio en particular. Este es el punto de partida para encontrar acceso a los diferentes tipos de datos generados por el proyecto en cuestión.
Los datos de RNA-Seq fueron generados para estudiar el rol del factor de transcripción SlyA en la resistencia de Salmonella a especies reactivas de oxígeno (ROS: reactive oxygen species), que son producidas por células fagocíticas del hospedero como mecanismo de defensa contra el patógeno.
Cabezas C.E., Briones A.C., Aguirre C., Pardo-Esté C., Castro-Severyn J., Salinas C.R., Baquedano M.S., Hidalgo A.A., Fuentes J.A., Morales E.H., Meneses C.A., Castro-Nallar E., Saavedra C.P. (2018). The transcription factor SlyA from Salmonella Typhimurium regulates genes in response to hydrogen peroxide and sodium hypochlorite. Research in Microbiology, 1–16.
Desde ahora en adelante vamos a llamar a los datos de secuenciación de genoma completo y de RNA-Seq: “referencia” y “experimento”, respectivamente.
Entonces, los datos experimentales (RNA-Seq) son:
| Muestra | Condición |
|---|---|
| WT control | cepa wild-type en condición control |
| WT H2O2 | cepa wild-type tratada con 1 mM H2O2 |
| ΔSlyA control | cepa mutant en condición control |
| ΔSlyA H2O2 | cepa mutant tratada con 1 mM H2O2 |
WT y ΔSlyA corresponden a Salmonella enterica subsp. enterica serovar Typhimurium. ΔSlyA es la cepa mutante por deleción del gen slyA. Las condiciones son: estrés oxidativo inducido por peróxido de hidrógeno (H2O2) y control.
14.2 Control de calidad de secuencias
El control de calidad de las secuencias o quality control es siempre el primer paso después de la secuenciación, aquí comienza el trabajo…
 In silico !
In silico !
In silico: Experimento biológico desarrollado en computador o vía simulación computacional.
![]()
El control de calidad (quality control) consiste, básicamente, en tres pasos principales:
- Evaluar las secuencias y definir los parámetros de filtro y trimming (si es que son necesarios).
- Realizar el filtro y trimming de los reads. <- paso más importante!
- Evaluar los reads que pasaron el control de calidad, para asegurar que está todo en orden con los datos que serán base del resto de tu estudio.
Filtrar y cortar (trim) son dos procesos diferentes que ocurren durante el procesamiento de las reads. Filtrar se refiere a eliminar una secuencia, en base a si cumple o no con cierto criterio de filtro (e.g., filtrar secuencias de largo menor a 50 pb). Mientras que, cortar se refiere a eliminar un cierto número de bases del extremo 5’ y/o 3’ de las reads según un valor umbral de calidad, proceso al que llamamos “trimming”.
La calidad de las secuencias se mide en el valor de Phred (Phred quality score). Durante la secuenciación, se asigna un valor de calidad a cada base de acuerdo a que tan confiable es la lectura. El valor de calidad de Phred o “Phred quality score” se representa con la letra “Q” y se puede interpretar como la probabilidad de que la base indicada sea incorrecta.
| Q | Probabilidad de que la base sea incorrecta | Exactitud |
|---|---|---|
| 10 | 1 en 10 | 90% |
| 20 | 1 en 100 | 99% |
| 30 | 1 en 1.000 | 99,9% |
| 40 | 1 en 10.000 | 99,99% |
Para la evaluación de las secuencias usamos FastQC. FastQC toma como input los reads en formato FASTQ y entrega estadísticas (i.e., número y largo de reads) y gráficos que muestran el estado de las secuencias en varios aspectos.
- Descarga e instala FastQC.
- Abre FastqQC y dirígete a
File->Open-> selecciona los archivos en formato fastq que quieras evaluar. - Espera.

FastQC
- Por favor, tómate unos minutos para explorar los módulos de FastQC. Te recomendamos hacerlo en conjunto con éste manual que explica qué es cada gráfico y cómo interpretarlo.
- Piensa en el criterio a aplicar para el procesamiento de las reads.
El criterio de procesamiento de las reads puede variar dependiendo de la naturaleza de los datos (i.e., ¿qué estás secuenciando?, ¿qué metodología de secuenciación?) y del objetivo del estudio (i.e., transcriptómica, búsqueda de variantes, construcción de genoma, etc.). Sin embargo, lo más importante es siempre deshacerte de las secuencias artefacto (i.e., adaptadores y partidores), reads y bases de baja calidad (quality score [QC] < 20 [1 de cada 100 bases podría ser incorrecta]), secuencias ambiguas (Ns), secuencias duplicadas (producto de la amplificación por PCR), secuencias sobrerrepresentadas, y secuencias muy pequeñas.
Puedes revisar el árbol de decisión de control de calidad en la imagen a continuación, para guiarte en los aspectos más importantes a considerar en éste paso.

Figure 3
Entonces, debemos hacer el control de calidad de las reads de la secuenciación del genoma completo de Salmonella (referencia) y del RNA-Seq (experimento).
Control de calidad raw data referencia:
Primero, para la evaluación de las secuencias usamos FastQC.
- Abre FastqQC y dirígete a
File->Open-> selecciona los archivos FASTQ que quieras evaluar de la referencia en/workshop_aqp/genomics/reference/raw_data_ref/ - Espera.

- Por favor, tómate unos minutos para explorar los módulos de FastQC. Te recomendamos hacerlo poniendo en práctica lo aprendido en la sección ?? - “Control de calidad de las secuencias”, o bien, en conjunto con éste manual.
- Piensa en el criterio a aplicar para el procesamiento de las reads.
Entonces, el criterio de filtro y trimming que vamos a aplicar es:
- Filtrar secuencias con un largo menor a 50 pb.
- Filtrar secuencias con Ns.
- Filtrar secuencias duplicadas.
- Filtrar secuencias de baja complejidad.
- Filtrar secuencias con una calidad promedio menor a 20.
- Cortar las primeras (5’) 10 bases de las secuencias.
- Cortar las bases de baja calidad (Q20) desde el extremo 3’ de las secuencias.
¿Qué son las secuencias de baja complejidad? Un read se considera de alta o baja complejidad de acuerdo a la composición de su secuenica. Secuencias altamente repetitivas se consideran simples, mientras que las secuencias menos repetitivas se consideran más complejas. Hay métodos disponibles para calcular la complejidad de las reads y filtrarlas de acuerdo a un valor umbral indicado por el usuario. En éste caso vamos a utilizar el método DUST, que asigna un valor de 0 a 100 de acuerdo a que tan frecuente ocurren diferentes trinucleótidos en la secuencia (ver más). Alto valor indica baja complejidad.
Ya que tenemos claro los parámetros de filtro y trimming, ¡vamos a procesar las reads!
Para procesar las reads, usamos PRINSEQ. Prinseq toma como input las secuencias en formato fastq, realiza primero el filtro y después el trimming de las reads según los parámetros escritos por el usuario, y finalmente, entrega archivos fastq con las secuencias procesadas.
- Recuerda que la forma de escribir la línea de comando de instrucciónes para Prinseq, o cualquier otro programa, es consultando las opciones del programa. Hay dos formas de hacerlo, seguir el manual, o bien, usar la opción
-ho-helpen la terminal:
$ prinseq-lite -h
Options:
-help | -h
Print the help message; ignore other arguments.
-man Print the full documentation; ignore other arguments.
-version
Print program version; ignore other arguments.
-verbose
Prints status and info messages during processing.
***** INPUT OPTIONS *****
-fastq <file>
Input file in FASTQ format that contains the sequence and
quality data. Use stdin instead of a file name to read from
STDIN (-fasta stdin). This can be useful to process compressed
files using Unix pipes.
...- Muévete a la carpeta en donde se encuentra el raw data de la referencia.
- Usa
prinseq-lite.plpara procesar el raw data.
# Corremos prinseq
$ perl prinseq-lite.pl -verbose -fastq ../raw_data_ref/reference_1.fastq -fastq2 ../raw_data_ref/reference_2.fastq -out_format 3 -out_good reference_OK -out_bad null -log reference.log -min_len 50 -min_qual_mean 20 -ns_max_n 1 -derep 1 -derep_min 6 -lc_method dust -lc_threshold 7 -trim_left 10 -trim_qual_right 20
# Fíjate en cómo le estamos entregando el input a prinseq
# Como los archivos no están en la carpeta actual, debemos anteponer la ruta a cada archivo
# Usamos '../' para ir una carpeta atrás, donde está la carpeta 'raw_data_ref/' y dentro de ella cada fastq
#
# Como tenemos paired reads ('-fastq' y '-fastq2'), prinseq genera dos archivos FASTQ con las single reads
# Las single reads o singletons son las secuencias provenientes de R1 o R2 que no pudieron ser apareadas después del trimming y filtrado de las reads
# Por razones prácticas, vamos a concatenar ambos singletons para tener solo uno
$ cat reference_OK_1_singletons.fastq reference_OK_2_singletons.fastq > reference_OK_singletons.fastq
$ rm reference_OK_1_singletons.fastq reference_OK_2_singletons.fastq- Recuerda que el último paso del control de calidad es revisar cómo resultó el procesamiento de las reads. Revisa cuánto dato se perdió producto del procesamiento. Mientras menor sea la diferencia entre el número de reads inicial vs. el número de reads procesados, mejor. En general, se recomienda no filtrar más del ~10% de tus datos, de lo contrario, quizás quieras relajar tu criterio de filtro.
# Prinseq, entre sus archivos de salida, entrega un reporte en formato con extensión '.log'
# Vamos a revisarlo para ver qué porcentaje de _reads_ se filtraron
$ cat reference.log
Estimate size of input data for status report (this might take a while for large files)
done
Parse and process input data
done
Clean up empty files
done
Input and filter stats:
Input sequences (file 1): 774,985
Input bases (file 1): 217,448,084
Input mean length (file 1): 280.58
Input sequences (file 2): 774,985
Input bases (file 2): 217,587,209
Input mean length (file 2): 280.76
Good sequences (pairs): 749,798
Good bases (pairs): 414,836,994
Good mean length (pairs): 553.27
Good sequences (singletons file 1): 19,611 (2.53%)
Good bases (singletons file 1): 5,717,905
Good mean length (singletons file 1): 291.57
Good sequences (singletons file 2): 536 (0.07%)
Good bases (singletons file 2): 149,737
Good mean length (singletons file 2): 279.36
Bad sequences (file 1): 5,576 (0.72%) # Porcentaje de secuencias filtradas desde el R1
Bad bases (file 1): 488,802
Bad mean length (file 1): 87.66
Bad sequences (file 2): 19,611 (2.53%) # Porcentaje de secuencias filtradas desde el R2
Bad bases (file 2): 5,845,442
Bad mean length (file 2): 298.07
Sequences filtered by specified parameters:
trim_qual_right: 374
min_len: 9019
min_qual_mean: 19723
ns_max_n: 712
lc_method: 399- Finalmente, puedes usar FastQC nuevamente para abrir los archivos FASTQ de la referencia después del procesamiento (
reference_OK_1.fastq,reference_OK_2.fastq), y explorar las diferencias (antes y después).
Control de calidad raw data experimento:
Repetimos la estrategia, usando FastQC y Prinseq para la evaluación y procesamiento de las reads producto de la secuenciación del transcriptoma de Salmonella bajo cuatro condiciones diferentes: WT control, WT H2O2, ΔSlyA control, y ΔSlyA H2O2. Tenemos 4 muestras que procesar: SlyA_control - SlyA_H2O2 - WT_control - WT_H2O2
De hecho, los autores del artículo asociado a estos datos ya hicieron este trabajo -> Miremos el artículo, específicamente la sección de materiales y métodos: 2 Materials and methods - 2.2. ROS treatment, RNA isolation, and RNA-seq analysis.


Cabezas C.E., Briones A.C., Aguirre C., Pardo-Esté C., Castro-Severyn J., Salinas C.R., Baquedano M.S., Hidalgo A.A., Fuentes J.A., Morales E.H., Meneses C.A., Castro-Nallar E., Saavedra C.P. (2018). The transcription factor SlyA from Salmonella Typhimurium regulates genes in response to hydrogen peroxide and sodium hypochlorite. Research in Microbiology, 1–16.
- Replicamos la línea de comando de Prinseq diseñada por los autores para hacer el control de calidad de las reads.
# Corremos prinseq para la réplica 1 de la muestra 'WT_control'
$ perl prinseq-lite.pl -verbose -fastq ../raw_data_exp/WT_control_1.fastq -fastq2 ../raw_data_exp/WT_control_2.fastq -out_format 3 -out_good WT_control_OK -out_bad null -log WT_control.log -min_len 50 -min_qual_mean 20 -trim_qual_left 20 -trim_qual_right 20 -ns_max_n 0 -lc_method dust -lc_threshold 7
# Como los archivos no están en la carpeta actual, debemos anteponer la ruta a cada archivo
# Usamos '../' para ir una carpeta atrás, donde está la carpeta 'raw_data_exp/' y dentro de ella cada fastq
#
# Por razones prácticas, vamos a concatenar ambos singletons para tener solo uno
$ cat WT_control_OK_1_singletons.fastq WT_control_OK_2_singletons.fastq > WT_control_OK_singletons.fastq
$ rm WT_control_OK_1_singletons.fastq WT_control_OK_2_singletons.fastqPuedes mirar el reporte de Prinseq que está escrito en el archvio con extensión
.log. Y también, usar FastQC para abrir los archivos FASTQ del experimento después del procesamiento (WT_control_OK_1.fastq,WT_control_OK_2.fastq), y explorar las diferencias (antes y después).Usa la misma línea de comando de Prinseq para procesar las 3 muestras restantes:
WT_H2O2-SlyA_control-SlyA_H2O2
14.3 Ensamble, anotación y evaluación
Antes de hacer un análisis de expresión diferencial de genes a partir de la secuenciación del transcriptoma (RNA-Seq) de un organismo bajo diferentes condiciones, necesitamos un genoma de referencia contra el cuál mapear las secuencias. Esto con el objetivo de identificar cuáles genes se están expresando y en qué abundancia. En este contexto, tenemos 2 opciones:
- Usar el genoma de referencia existente del organismo. Generalmente, se prefiere usar el genoma de referencia disponible, especialmente en el caso de especies modelo, para las cuáles se ha hecho un gran esfuerzo en ensamblar y curar sus genomas liberando nuevas y mejoradas versiones cada cierto tiempo. Es necesario tener en cuenta que el uso de un genoma de referencia viene con una limitación que será importante o no dependiendo del objetivo o pregunta de tu estudio. Al usar un genoma de referencia te limitas a observar la información que este contiene; si tu organismo en estudio tiene alguna variación en particular, como una inserción que no está representada en la referencia, no serás capaz de verlo en el análisis.
- Una excelente forma de averigüar si tu organismo de interés tiene o no un genoma de referencia publicadol, es consultando las bases de datos. Dirígete a la base de datos de genomas de NCBI: Genome -> escribe Salmonella enterica en la barra de búsqueda -> haz clic en
Search.

Toma unos minutos para explorar la página. La base de datos RefSeq de NCBI, hace un esfuerzo en clasificar los genomas -que científicos de todas partes del mundo suben a las bases de datos- en diferentes categorías según su calidad. Fíjate que hay más de 14.600 genomas disponibles para la especie Salmonella enterica, sin embargo, existe un genoma de referencia: “Reference genome: Salmonella enterica subsp. enterica serovar Typhi str. CT18”. Observa también, la sección Representative (genome information for reference and representative genomes) en la misma página. Para conocer más acerca de los genomas procariontes de referencia y representativos dirígete a: Prokaryotic RefSeq Genomes.
- La otra opción, es secuenciar y hacer un ensamble de novo del genoma, para posteriormente usarlo como genoma de referencia. Estrategia muy utilizada para el estudio de organismos no modelo, de nuevos aislados bacterianos, etc.
A pesar de que contamos con un genoma de referencia para Salmonella enterica subsp. enterica serovar Typhimurium, para efectos del práctico vamos a hacer el ensamble de novo del genoma.
14.3.1 Ensamble de novo del genoma de Salmonella enterica subsp. enterica serovar Typhimurium
Como mencionamos antes en la sección “3.1 Set de datos”, vamos a utilizar el raw data de la secuenciación del genoma completo de la especie alojados en el BioProject Accession: PRJNA280335. Por favor, haz clic en el link y en la tabla de contenidos que aparece en la parte superior derecha de la página: Related information -> haz clic en SRA. Esto debería llevarte a la base de datos SRA de NCBI que aloja el raw data e información sobre el proceso de secuenciación.


En esta oportunidad, nosotros ya hemos descargado y procesado las reads que necesitamos. Pero, si en un futuro necesitas descargar el raw data de algún estudio de tu interés, puedes ir al BioProject correspondiente y buscar el link al SRA. Puedes econtrar el link de acceso en la sección de materiales y métodos del artículo ciéntifico de tu interés, o bien, usando palabras claves en la barra de búsqueda de NCBI. Finalmente, puedes usar NCBI SRA Toolkit para bajar los archivos FASTQ de SRA.
Teniendo las reads procesadas, podemos hacer el ensamble de novo. Para ello, vamos a usar el algoritmo para ensamble de genoma SPAdes (PMID: 24093227, PMID: 22506599).
Sigue las instrucciones de instalación de SPAdes según tu sistema operativo aquí.
SPAdes necesita como input (archivo de entrada) el o los archivos FASTQ o FASTA con las secuencias o reads que se quieren ensamblar. En este caso, nuestro input para SPAdes es el output (archivo de salida o resultado) de Prinseq.
# Corremos spades
spades.py -1 ../qc_ref/reference_OK_1.fastq -2 ../qc_ref/reference_OK_2.fastq -s ../qc_ref/reference_OK_singletons.fastq -t 16 -m 64 -o spades_out -k 21,33,55,77,99,127 --careful --cov-cutoff auto
# Como los archivos no están en la carpeta actual, debemos anteponer la ruta a cada archivo
# Usamos '../' para ir una carpeta atrás, donde está la carpeta 'qc_ref/' y dentro de ella cada fastq
# La opción '-o' es para indicar la carpeta de salida: el output de spades se guarda en spades_out/
# Una vez finalizado el ensamble, mira la salida de spades
$ cd spades_out/
$ ls
K127/ before_rr.fasta mismatch_corrector/
K21/ contigs.fasta params.txt
K33/ contigs.paths scaffolds.fasta
K55/ contigs_over200bp.txt scaffolds.paths
K77/ corrected/ spades.log
K99/ dataset.info tmp/
assembly_graph.fastg input_dataset.yaml
assembly_graph_with_scaffolds.gfa misc/
# Como puedes ver, vas a encontrar muchos archivos
# Lo que nos interesa aquí es el genoma que acabamos de reconstruir a partir del ensamble de las reads producto de la secuenciación del ADN aislado de un cultivo de Salmonella enterica
# Esto sería el archivo que guarda los contigs en formato fasta: 'contigs.fasta'Descripción de comando: Usa spades.py -h para ver las opciones disponibles y sus respectivas descripciones en la terminal, o bien, revisa el manual.
-k 21,33,55,77,99,127: La opción -k es para indicar los valores de tamaño de k-mer a utilizar, en este caso seguimos la recomendación indicada en el manual for assembling long Illumina paired reads, porque nuestro input son paired reads de 300pb (2x300) de Illumina.
14.3.2 Anotación del genoma de Salmonella enterica subsp. enterica serovar Typhimurium
Al realizar el ensamble de los metagenomas, lo que obtenemos son cientos de miles de contigs, que son una representación, más bien fragmentada, de los genomas de todos los microorganismos presentes en la muestra. Cada contig, puede contener uno o más genes codificados en su secuencia. Para acceder a éstos genes, primero, es necesario identificarlos dentro de las secuencias, básicamente, mediante la búsqueda de marcos abiertos de lectura. Desde la anotación estructural, se obtienen las coordenadas (i.e., posiciones en la secuencia) de los genes y sus regiones codificantes (CDSs [coding sequences]), las que son traducidas para generar las secuencias aminoacídicas (proteínas). Por otra parte, la anotación funcional consiste en resolver qué función cumplen éstos genes o para qué proteína codifican. Ésto, mediante la comparación de las secuencias nucleotídicas y/o aminoacídicas predichas, contra bases de datos (e.g., NCBI nt y nr, UniProt, etc.), en donde la secuencia problema “hereda” la función de su secuencia homóloga en la base de datos.
Prokka (rapid prokaryotic genome annotation; PMID: 24642063) es un programa para anotar genomas procariotas, popular por su rapidez y archivos de salida útiles para posteriores análisis (e.g., fastas con proteínas y genes, archivos de anotación GFF3 y GBK, etc.).
- Sigue las instrucciónes de instalación de Prokka aquí.
- Prokka necesita como input un archivo FASTA con los contigs ensamblados.
# Corremos prokka
perl prokka --outdir prokka_out --prefix STyphimurium --locustag STy --genus Salmonella --species enterica --kingdom Bacteria --gram neg --cpus 12 --evalue 1e-5 ../assembly_ref/reference.fasta
# La opción '--outdir' es para indicar la carpeta de salida: el output de prokka se guarda en prokka_out/
# La opción `--prefix` es para definir el prefijo de los archivos de salida
# La opción `--locustag` es para definir el prefijo del nombre de cada locus anotado en el genoma de Salmonella
# Una vez finalizada la anotación, miramos la salida de prokka
$ cd prokka_out/
$ ls
STyphimurium.err STyphimurium.ffn STyphimurium.fsa STyphimurium.gff STyphimurium.sqn STyphimurium.tsv
STyphimurium.faa STyphimurium.fna STyphimurium.gbk STyphimurium.log STyphimurium.tbl STyphimurium.txtDescripción de comando: Usa prokka -h para ver las opciones disponibles y sus respectivas descripciones en la terminal, o bien, revisa el manual.
Prokka genera varios archivos de salida que contienen todo tipo de información acerca de las características del genoma. Las que vamos a necesitar a continuación son gff y faa.
14.3.3 Evaluación del genoma
Vamos a evaluar el ensamble en dos niveles. Primero, a través del cálculo de estadísticas a partir de las secuencias ensambladas. Segundo, a través de la búsqueda de genes ortólogos altamente conservados en genomas bacterianos a partir de los CDS predichos en el genoma ensamblado.
14.3.3.1 Estadísticas de secuencias
Las estadísticas a calcular son el número de secuencias o contigs, número total e individual de bases (A, T, C, G, y la base ambigua N), porcentaje de G+C y A+T, largo mínimo, máximo, promedio, y N50. El valor de N50 es el largo mínimo de contig necesario para cubrir el 50% del genoma con contigs de largo igual o mayor a este valor. Mayor el valor de N50, mejor el ensamble.
Estas estadísticas nos dan cuenta del tamaño del genoma ensamblado (largo total o número total de bases), del porcentaje de GC, y de qué tan fragmentado está. Este último se refiere al número y tamaño de los contigs, buscamos tener el menor número de contigs posible y que estos sean largos, es más, lo ideal es poder ensamblar un contig por cromosoma. En el caso de bacterias, por ejemplo, un genoma completo consta de una secuencia del largo total del genoma, quizás acompañada de una segunda secuencia correspondiente a un plásmido.

Vamos a usar NGS QC Toolkit (PMID: 22312429), un compilado de herramientas para hacer control de calidad de datos NGS. En este caso vamos a usar N50Stat.pl, este programa toma como input un archivo en formato FASTA y calcula diferentes estadísticas.
- Descarga NGSQCToolkit aquí.
- Usamos
N50Stat.plpara analizar el ensamble.
# Corremos N50Stat.pl
$ perl ~/programs/NGSQCToolkit/Statistics/N50Stat.pl -i reference.fasta -o reference_STATSreport
# La opción '-i' es para indicar el archivo input
# La opción '-o' es para indicar el nombre del archivo output
# Una vez terminado, mira el resultado
$ cat reference_STATSreport
Total sequences 124
Total bases 4782820
Min sequence length 208
Max sequence length 403178
Average sequence length 38571.13
Median sequence length 1698.00
N25 length 231338
N50 length 113211
N75 length 65198
N90 length 33736
N95 length 26606
As 23.78 %
Ts 24.00 %
Gs 26.34 %
Cs 25.88 %
(A + T)s 47.78 %
(G + C)s 52.22 %
Ns 0.00 %
# Quizás, deberíamos ponerlo en una tabla| Description | Value |
|---|---|
| Total sequences | 124 |
| Total bases | 4.78 Mb |
| Min sequence length | 208 |
| Max sequence length | 403,178 |
| Average sequence length | 38,571 |
| Median sequence length | 1,698 |
| N25 length | 231,338 |
| N50 length | 113,211 |
| N75 length | 65,198 |
| N90 length | 33,736 |
| N95 length | 26,606 |
| As | 23.78 % |
| Ts | 24.00 % |
| Gs | 26.34 % |
| Cs | 25.88 % |
| (A + T)s | 47.78 % |
| (G + C)s | 52.22 % |
| Ns | 0.00 % |
¿Cómo saber si estos valores son buenos o no? Comparándolos con los valores calculados en los otros más de 14.500 genomas de Salmonella enterica alojados en la base de datos Genome de NCBI. Mira los valores en la sección Summary de la página del genoma, la misma que hemos estado mirando (ve la imagen arriba). ¿Son los valores similares?
14.3.3.2 Análisis de completitud
El análisis de completitud, se basa en que si el genoma ha sido secuenciado y ensamblado completamente debería al menos contener todos los genes ortólogos altamentes conservados en el Kingdom Bacteria, por ejemplo, si es que estamos ensamblando un genoma bacteriano.
Para analizar la completitud del genoma ensamblado, vamos a usar BUSCO (PMID: 29220515, PMID: 26059717). A partir de la anotación del genoma ensamblado, BUSCO identifica la presencia en copia única de genes ortólogos altamente conservados, según organismo, descritos en la base de datos OrthoDB.
- Descarga e instala BUSCO aquí.
- Usamos BUSCO para evaluar la completitud del genoma a partir de las regiones codificantes predichas.
# Corremos BUSCO
$ python run_BUSCO.py -i prokka_out/STyphimurium.faa -c 8 -o STyphimurium -m prot -l /busco/datasets/bacteria_odb9
# Como los archivos no están en la carpeta actual, debemos anteponer la ruta a cada archivo
# Usamos 'prokka_out/' para acceder al archivo 'STyphimurium.faa'
# BUSCO genera varios archivos de salida, esta vez nos enfocamos en el archivo 'run_STyphimurium/short_summary_STyphimurium.txt'
# Mira el resultado
$ cat run_STyphimurium/short_summary_STyphimurium.txt
BUSCO version is: 3.0.2
The lineage dataset is: bacteria_odb9 (Creation date: 2016-11-01, number of species: 3663, number of BUSCOs: 148)
To reproduce this run: python /home/ecastron/programs/busco/scripts/run_BUSCO.py -i ../prokka_out/STyphimurium.faa -o STyphimurium -l /home/ecastron/programs/busco/datasets/bacteria_odb9/ -m proteins -c 8
Summarized benchmarking in BUSCO notation for file ../prokka_out/STyphimurium.faa
BUSCO was run in mode: proteins
C:98.6%[S:98.6%,D:0.0%],F:0.0%,M:1.4%,n:148
146 Complete BUSCOs (C)
146 Complete and single-copy BUSCOs (S)
0 Complete and duplicated BUSCOs (D)
0 Fragmented BUSCOs (F)
2 Missing BUSCOs (M)
148 Total BUSCO groups searchedDescripción de comando: Usa python run_BUSCO.py -h para ver las opciones disponibles y sus respectivas descripciones en la terminal, o bien, revisa el manual.
El genoma ensamblado muestra un alta completitud, con un 98.6% de los genes ortólogos altamente conservados en bacterias, descritos en la base de datos OrthoDB.
Se considera aceptable un valor de completitud mayor o igual al 85% (PMID: 23870653).
Vamos a actualizar la tabla de estadísticas del ensamble del genoma de Salmonella enterica subsp. enterica serovar Typhimurium.
- Agregamos el número de proteínas predichas y los resultados del análisis de completitud.
| Description | Value |
|---|---|
| Total sequences | 124 |
| Total bases | 4.78 Mb |
| Min sequence length | 208 |
| Max sequence length | 403,178 |
| Average sequence length | 38,571 |
| Median sequence length | 1,698 |
| N25 length | 231,338 |
| N50 length | 113,211 |
| N75 length | 65,198 |
| N90 length | 33,736 |
| N95 length | 26,606 |
| As | 23.78 % |
| Ts | 24.00 % |
| Gs | 26.34 % |
| Cs | 25.88 % |
| (A + T)s | 47.78 % |
| (G + C)s | 52.22 % |
| Ns | 0.00 % |
| Predicted proteins | 4,430 |
| Proteome completeness | C:98.6%[S:98.6%,D:0.0%],F:0.0%,M:1.4%,n:148 |
“Proteome completness”: C = complete orthologs, S = complete and single-copy orthologs, D = complete and duplicated orthologs, F = fragmented orthologs, M = missing orthologs.
- Compara el número de proteínas predichas en el genoma ensamblado con el número de proteínas en los otros más de 14.500 genomas de Salmonella enterica alojados en la base de datos Genome de NCBI. ¿Similar?
14.4 Transcriptómica
Transcriptómica es el estudio del transcriptoma, que corresponde al conjunto de transcritos/ARN generados a partir del genoma, bajo diferentes condiciones. Mediante la comparación de transcriptomas, provenientes de diferentes muestras, podemos identificar genes que se expresan de forma diferencial bajo las diferentes condiciones o tratamientos bajo lo cuáles estaban las muestras, y asociarlos a una función o proceso.
El análisis in silico de un experimento de RNA-Seq comienza con un conjunto de archivos FASTQ que contienen las secuencias nucleotídicas de los transcritos. Estas secuencias deben ser alineadas a un genoma o transcriptoma de referencia, para luego contar el número de reads existentes por cada gen o región codificante y así estimar su nivel de expresión transcripcional en la muestra.

Figure 10 Mapping reads to a reference or de novo assembly. Reprinted by permission from Macmillan Publishers Ltd: Nature Biotechnology. Haas BJ and Zody MC. Advancing RNA-seq analysis. 28:421-423, copyright 2010 (10). Fuente: Read mapping or alignment
14.4.1 Alineamiento de reads en contra del genoma de referencia
Mapping reads se refiere al alineamiento de las reads (secuencias cortas producto de la secuenciación) en contra de secuencias de referencia (secuencias largas que representan un genoma o transcriptoma). Vamos a alinear las reads de cada una de las muestras (SlyA_control, SlyA_H2O2, WT_control, WT_H2O2) por separado, en contra del genoma de referencia de de Salmonella enterica subsp. enterica serovar Typhimurium que recién generamos.
Vamos a usar Bowtie2 (PMID: 22388286), un algoritmo diseñado para el alineamiento de reads en contra de largas secuencias de referencia.
- Sigue las instrucciones de instalación de Bowtie2 aquí.
- Usamos Bowtie2 para alinear las reads de cada una de las 8 muestras por separado en contra del genoma de referencia:
- Primero, necesitamos indexar la referencia usando
bowtie2-build - Segundo, corremos el alineamiento usando
bowtie2
- Primero, necesitamos indexar la referencia usando
# Crea una nueva carpeta llamada "mapping".
$ mkdir mapping
$ cd mapping/
# Crea una nueva carpeta llamada "bt2_index", aquí vamos a indexar la referencia
$ mkdir bt2_index
$ cd bt2_index/
# Corremos bowtie2-build para construir el bt2 index (index de bowtie2)
bowtie2-build reference.fasta reference
# 'reference' es el prefijo que tendrán todos los archivos output de bowtie2-build
# Una vez finalizado, mira la salida de bowtie2-build
$ ls -1
reference.1.bt2
reference.2.bt2
reference.3.bt2
reference.4.bt2
reference.rev.1.bt2
reference.rev.2.bt2
# Todos estos archivos son los índices (index) de la referencia que bowtie2 va a utilizar para mapear las reads
# Muévete a la carpeta 'mapping' y corre bowtie2
$ cd ../
# Comenzamos con la muestra: WT_control_rep1
bowtie2 --end-to-end --sensitive -p 24 -x bt2_index/reference -1 WT_control_OK_1.fastq -2 WT_control_OK_2.fastq -U WT_control_OK_singletons.fastq -S WT_control.samDescripción de comando: Usa bowtie2-build -h/bowtie2 -h para ver las opciones disponibles y sus respectivas descripciones en la terminal, o bien, revisa el manual.
- Usa la misma línea de comando de Bowtie2 para alinear las otras 3 muestras restantes en contra del genoma de referencia:
WT_H2O2-SlyA_control-SlyA_H2O2
Para entender mejor el proceso de indexar la referencia y de mapeo de reads en contra de secuencias largas de referencia, mira esta página de Biostars y este artículo.
- Una vez realizado el mapeo de todas la muestras, mira la salida de Bowtie2.
$ ls -1
SlyA_control.sam
SlyA_H2O2.sam
WT_control.sam
WT_H2O2.samEl output del alineamiento es guardado en un archivo formato SAM/BAM, SAM por Sequence Alignment/Map y BAM es el equivalente en binario. Este formato de archivo guarda la información de dónde y cómo cada read se alinea en la referencia.
14.4.2 Conteo de reads
El siguiente paso, es calcular el número de reads alineados a cada región codificante o CDS identificados en el proceso de anotación en el genoma de referencia. El objetivo, es obtener una tabla con el número de reads por CDS en cada muestra.
Para contar el número de reads por CDS vamos a utilizar HTSeq (PMID: 25260700), un programa que cuenta con varias herramientas para el análisis de datos HTS (high-throughput sequencing), incluyendo HTSeq-count. A partir del output del alineamiento (archivo SAM/BAM) y del output de la anotación (archivo GFF), HTSeq-count es capaz de contar las reads alineadas a cada CDS en el genoma de referencia.
HTSeq-count requiere que el archivo SAM/BAM incluya los alineamientos de las paired reads solamente y que esté ordenado por nombre de las reads. Samtools es un conjunto de herramientas para el manejo de archivos de alineamiento en formato SAM/BAM.
- Descarga e instala Samtools aquí.
- Usamos
samtools viewysamtools sortpara configurar los archivos SAM de las muestras.
# Corremos samtools
samtools view -hf 1 WT_control.sam -@ 24 | samtools sort -n -o WT_control_nsort.sam -@ 24
# La opción '-@' es para indicar el número de threads a utilizar
# Usamos '|' para concatenar 'samtools view' y 'samtools sort'
# Mira el resultado
$ ls -1
SlyA_control_nsort.sam
SlyA_control.sam
SlyA_H2O2_nsort.sam
SlyA_H2O2.sam
WT_control_nsort.sam
WT_control.sam
WT_H2O2_nsort.sam
WT_H2O2.sam
# Ahora tenemos los archivos SAM output del alineamiento y también los que configuramos con samtoolsDescripción de comando: Usa samtools view -h/samtools sort -h para ver las opciones disponibles y sus respectivas descripciones en la terminal, o bien, revisa el manual.
samtools view -hf 1 : Las opciones -hf 1 son para indicar que al leer el archivo SAM se deben agregar los headers (títulos de columna) y considerar las paired reads que alinearon solamente.
samtools sort -n : Indica que el archivo SAM debe ser ordenado según el nombre de las reads (-n).
Usa la misma línea de comando de Samtools para configurar los archivos SAM de las otras 3 muestras restantes:
WT_H2O2_rep1-SlyA_control-SlyA_H2O2Ahora que tenemos los inputs necesarios: archivos SAM configurados y archivo GFF. Procedemos a usar HTSeq-count.
# Ccrea una nueva carpeta llamada 'read_counts'
$ mkdir read_counts
$ cd read_counts/
# Configura el archivo GFF para quitar la sección '#FASTA' que HTSeq-count no reconoce
# Usamos el comando 'vi' para escribir un nuevo archivo copiando un rango de líneas específicas del archivo 'STyphimurium.gff'
$ vi STyphimurium.gff
# Una vez abierto el archivo, escribe:
:1,5072w STyphimurium_ref.gff
# Esta instrucción indica que se debe copiar desde la línea 1 hasta la línea 5072 del archivo 'STyphimurium.gff' en un nuevo archivo llamado 'STyphimurium_ref.gff'
# Cuando aparezca el mensaje "STyphimurium_ref.gff written", puedes salir del editor escribiendo:
:q!
# El nuevo archivo 'STyphimurium_ref.gff' quedará guardado en la carpeta actual
# Corremos htseq-count
htseq-count --stranded=no --format=sam --order=name --type=CDS --idattr=locus_tag --mode=intersection-nonempty WT_control_nsort.sam STyphimurium_ref.gff > WT_control_counts.tsv
# Mira el output de htseq-count
$ head -n5 WT_control_counts.tsv
STy_00001 60
STy_00002 1
STy_00003 9
STy_00004 850
STy_00005 25
# La primera columna contiene los nombres asignados a cada CDS en el proceso de anotación
# La segunda columna contiene el número de reads por cada CDS en la muestra 'WT_control'Descripción de comando: Usa htseq-count para ver las opciones disponibles y sus respectivas descripciones en la terminal, o bien, revisa el manual.
Al correr htseq-count usamos las opciones --order=name --type=CDS --idattr=locus_tag --additional-attr=gene para indicar que: los archivos SAM están ordenados por nombre de las reads, que se deben contar las reads por CDS en la referencia, y que se deben indicar el atributo locus_tag. Los atributos están descritos en el archivo GFF para cada CDS, locus_tag es el nombre del locus.
La opción --mode=intersection-nonempty es para indicar el modo en que htseq-count va a considerar el alineamiento de cada read como válido o no para contarlo. La siguiente figura muestra los efectos de los tres modos disponibles:
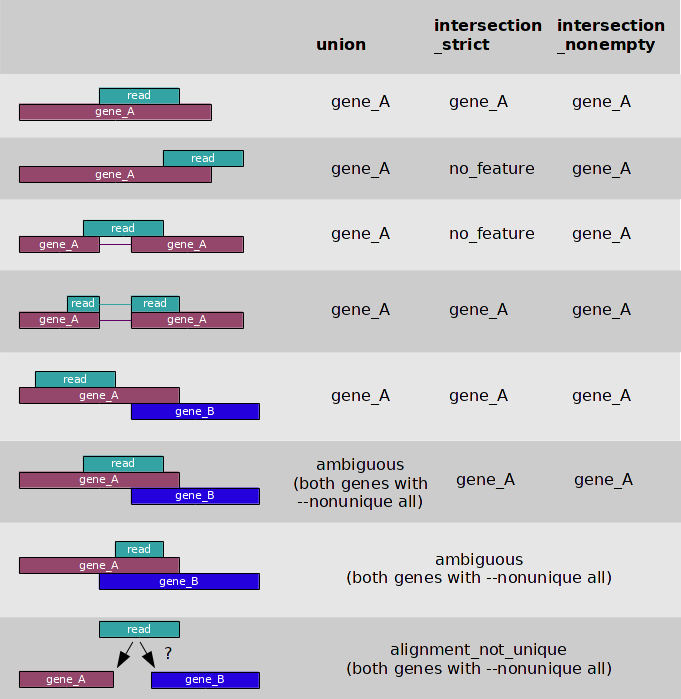
En nuestro caso usamos el modo intersection-nonempty considerando que en los genomas bacterianos ocurre la superposición de regiones codificantes.
- Usa la misma línea de comando de Samtools para configurar los archivos SAM de las otras 3 muestras restantes:
WT_H2O2-SlyA_control-SlyA_H2O2
Teniendo las tablas de cuentas de reads para cada CDS en el genoma en las 3 muestras, podemos pasar al siguiente paso: análisis de expresión diferencial.
14.4.3 Análisis de expresión diferencial de genes
Contamos con una tabla que muestra el número de reads (valores) por CDS (filas) anotado en el genoma de referencia de Salmonella enterica subsp. enterica serovar Typhimurium, por cada una de las 4 muestras (columnas): WT_control - WT_H2O2 - SlyA_control - SlyA_H2O2. Este es un punto de transición, en el que pasamos de la terminal a R. Usamos R para leer las tablas de cuentas y hacer el análisis de expresión diferencial usando el paquete DESeq2 (PMID: 25516281).
# Para hacer el análisis de expresión diferencial, necesitamos que la tabla sea de la siguiente forma:
# Filas: locus
# Columnas: muestras14.4.3.1 Transición a R
Lo primero que tenemos que hacer es instalar y cargar los paquetes que vamos a usar en esta sección. Algunos están alojados en el repositorio CRAN y otros en el repositorio Bioconductor.
# Generamos objetos con la lista de paquetes que queremos instalar y cargar
cran_packages <- c("knitr", "qtl", "bookdown", "magrittr", "plyr", "ggplot2", "grid", "gridExtra", "tidyverse", "devtools", "dplyr", "pheatmap", "RColorBrewer", "PoiClaClu", "gtools")
bioc_packages <- c("airway","DESeq2", "genefilter", "GenomeInfoDb")
# Cargar paquetes
sapply(c(cran_packages, bioc_packages), require, character.only = TRUE)## knitr qtl bookdown magrittr plyr ggplot2
## TRUE TRUE TRUE TRUE TRUE TRUE
## grid gridExtra tidyverse devtools dplyr pheatmap
## TRUE TRUE TRUE TRUE TRUE TRUE
## RColorBrewer PoiClaClu gtools airway DESeq2 genefilter
## TRUE TRUE TRUE TRUE TRUE TRUE
## GenomeInfoDb
## TRUE- Como tenemos múltiples outputs de HTSeq-count, usamos la función ‘DESeqDataSetFromHTSeqCount’ para leer las tablas de conteo por muestra y combinarlas en una.
# Define el directorio en donde se encuentra el output de htseq-count
directory <- "/Users/ecastron/Dropbox/10_courses/2020/MCV502_BCM634_BIO625/labs/data/"
# Definimos los archivos a leer usando 'list.files' y seleccionamos los archivos que contienen "counts" usando grep. Usamos la función 'sub' para sacar el nombre de las muesras a partir del nombre de los archivos.
sampleFiles <- grep("counts",list.files(directory),value=TRUE)
sampleCondition <- sub("(.*_counts).*","\\1",sampleFiles)
sampleTable <- data.frame(sampleName = sampleFiles,
fileName = sampleFiles,
condition = sampleCondition)
# Usamos la función 'DESeqDataSetFromHTSeqCount' para crear el objeto DESeq
dds <- DESeq2::DESeqDataSetFromHTSeqCount(sampleTable = sampleTable,
directory = directory,
design= ~ condition)## Warning in DESeqDataSet(se, design = design, ignoreRank): some variables in
## design formula are characters, converting to factors## class: DESeqDataSet
## dim: 4430 4
## metadata(1): version
## assays(1): counts
## rownames(4430): STy_00001 STy_00002 ... STy_04509 STy_04514
## rowData names(0):
## colnames(4): SlyA_control_counts.tsv SlyA_H2O2_counts.tsv
## WT_control_counts.tsv WT_H2O2_counts.tsv
## colData names(1): condition- Revisamos el objeto DESeq2 creado:
## SlyA_control_counts.tsv SlyA_H2O2_counts.tsv WT_control_counts.tsv
## STy_00001 212 179 60
## STy_00002 1 2 1
## STy_00003 65 116 9
## WT_H2O2_counts.tsv
## STy_00001 170
## STy_00002 25
## STy_00003 315## DataFrame with 4 rows and 1 column
## condition
## <factor>
## SlyA_control_counts.tsv SlyA_control_counts
## SlyA_H2O2_counts.tsv SlyA_H2O2_counts
## WT_control_counts.tsv WT_control_counts
## WT_H2O2_counts.tsv WT_H2O2_counts- Antes de continuar, vamos a hacer algunas modificaciones:
- Primero, vamos a renombrar los nombres de las columnas de la tabla de cuentas
- Vamos a agregar la tabla de información acerca de las muestras que hemos preparado una, descárgala aquí.
# Exportamos la tabla de cuentas desde el objeto DESeq a un nuevo objeto de tipo data.frame
countdata <- assay(dds)
countdata <- as.data.frame(countdata)
# Modificamos el nombre de las columnas
colnames(countdata) <- c("SlyA_control", "SlyA_H2O2", "WT_control", "WT_H2O2")
# Lee la tabla de información de las muestras que preparamos
coldata <- read.table(file = "/Users/ecastron/Dropbox/10_courses/2020/MCV502_BCM634_BIO625/labs/data/sample_information.tsv", sep = "\t", header = TRUE, row.names = 1, stringsAsFactors = TRUE)
coldata## strain condition
## SlyA_control SlyA control
## SlyA_H2O2 SlyA H2O2
## WT_control WT control
## WT_H2O2 WT H2O2- Ahora, tenemos todos los ingredientes para preparar un objeto adecuado para el análisis, llamados:
- countdata: tabla con las cuentas
- coldata: tabla con la información acerca de las muestras
- Estamos listos para construir el objeto DESeqDataSet a partir de countdata y coldata.
# Objeto DESeqDataSet
dds <- DESeqDataSetFromMatrix(countData = countdata,
colData = coldata,
design = ~ strain + condition)
dds## class: DESeqDataSet
## dim: 4430 4
## metadata(1): version
## assays(1): counts
## rownames(4430): STy_00001 STy_00002 ... STy_04509 STy_04514
## rowData names(0):
## colnames(4): SlyA_control SlyA_H2O2 WT_control WT_H2O2
## colData names(2): strain conditionEl argumento design de la función DESeqDataSetFromMatrix() es para indicar el diseño experimental, i.e., strain y condition, que corresponden a la cepa control (WT) o mutante (SlyA) y a si es que se trataron con peróxido de hidrógeno (H2O2) o no (control), respectivamente.
La fórmula ~ strain + condition es interpretada como: la diferencia en expresión génica según tratamiento condition tomando en cuenta si la cepa es wild-type o mutante strain.
Para los siguientes pasos utilizamos en gran medida el artículo de F1000 research sobre análisis de expresión génica. Existen otros métodos de cómo hacer un análisis de expresión génica y para escoger conviene revisar la literatura en busca de estudios de benchmark donde se comparan distintos métodos para identificar las bondades y limitaciones de cada uno. El objetivo no es nunca encontrar un vencedor sino que más bien identificar en qué caso o con qué tipo de datos cada método funciona mejor. Un ejemplo de estos estudios lo pueden encontrar aquí.
- Revísemeos rápidamente cuántas reads o fragmentos mapearon en contra de cada muestra (en millones de reads).
## SlyA_control SlyA_H2O2 WT_control WT_H2O2
## 6.2 5.8 6.9 6.814.4.3.2 Prefiltrado y algunas transformaciones
En el objeto DESeq las filas de dds corresponden a los genes y las columnas a las muestras.
- Para contar el número de genes simplemente ejecutamos:
## [1] 4430Ahora, en nuestros datos hay muchos genes que contienen cero reads y que no los necesitamos para nuestro análisis.
- Eliminamos aquellas filas que contengan genes con cero número de reads.
## [1] 4373- ¿Por qué algunos genes tienen 0 reads? ¿Qué significa esto?
[?]Muchos métodos estadísticos que comúnmente se usan en el análisis de expresión génica como PCA o clustering dependen de que los datos sean homocedásticos, i.e., que la varianza para cada gen tenga la misma distancia de la media. Sin embargo, normalmente los datos de RNA-seq no se comportan así sino que la varianza aumenta con la media. Ocurre que la mayor varianza observada depende de unos pocos genes que muestran las diferencias absolutas más grandes entre las muestras. Una simple y muy usada estrategia para evitar este problema, es calcular el logaritmo de las cuentas normalizadas; sin embargo, ahora los genes con cuentas bajas tenderán a dominar los resultados porque, el logaritmo tiende a amplificar las diferencias para los valores más pequeños. Es por esto que necesitamos transformar los datos, DESeq2 ofrece transformar las cuentas para estabilizar la varianza a través de la media.
- Usamos la
regularized-logarithmic transformationde la siguiente manera.
## SlyA_control SlyA_H2O2 WT_control WT_H2O2
## STy_00001 7.768907 7.342574 6.531974 6.970530
## STy_00002 1.831751 1.889341 1.838066 2.782111
## STy_00003 6.282234 6.627743 4.744976 7.354653- Vamos a comparar esta transformación con otra. Calculamos log2 y graficamos los resultados lado a lado.
par( mfrow = c( 1, 2 ) )
dds <- estimateSizeFactors(dds)
plot(log2(counts(dds, normalized=TRUE)[,1:2] + 1),
pch=16, cex=0.3)
plot(assay(rld)[,1:2],
pch=16, cex=0.3)
- ¿Qué conclusión se desprende del gráfico?
[?]14.4.3.3 Distancias entre muestras
Como un control útil antes de hacer un análisis diferencial de genes, es bueno evaluar la similitud global entre muestras. ¿Qué muestras son similares o diferentes entre sí? ¿La similitud entre las muestras se ajusta a las expectativas (controles con controles, tratamiento con tratamiento)?
## SlyA_control SlyA_H2O2 WT_control
## SlyA_H2O2 63.18370
## WT_control 48.70190 75.05033
## WT_H2O2 65.31942 33.59187 71.39640library("pheatmap")
library("RColorBrewer")
sampleDistMatrix <- as.matrix( sampleDists )
rownames(sampleDistMatrix) <- paste( rld$strain, rld$condition, sep = " " )
colnames(sampleDistMatrix) <- NULL
colors <- colorRampPalette( rev(brewer.pal(9, "Blues")) )(255)
pheatmap(sampleDistMatrix,
clustering_distance_rows=sampleDists,
clustering_distance_cols=sampleDists,
col=colors)
El mapa de calor o heatmap nos muestra qué muestras se parecen entre sí. En este caso, las muestras se agrupan de acuerdo a si recibieron tratamiento o no. Dependiento del tipo de medida de distancia, podríamos conseguir mayor o menor resolución al comparar las muestras. Probemos con otra medida de distancia. En vez de distancia euclideana vamos a usar la distancia de Poisson.
library("PoiClaClu")
poisd <- PoissonDistance(t(counts(dds)))
samplePoisDistMatrix <- as.matrix( poisd$dd )
rownames(samplePoisDistMatrix) <- paste( rld$strain, rld$condition, sep=" " )
colnames(samplePoisDistMatrix) <- NULL
pheatmap(samplePoisDistMatrix,
clustering_distance_rows=poisd$dd,
clustering_distance_cols=poisd$dd,
col=colors)
14.4.3.4 Análisis de componentes principales
Otra manera de visualizar las distancias entre las muestras es a través de un análisis de componentes principales o PCA. La idea es que las muestras sean proyectadas en un plano 2D de manera tal de maximizar las diferencias entre ellas. Por definición, el eje X en un PCA es la dirección que separa las muestras mayormente. En nuestro caso “dirección” se refiere a una combinación de genes. Veamos esto en acción.

También podríamos tomar los datos con que se construyó este gráfico y usarlos para construir un gráfico más personalizado en ggplot2.
## PC1 PC2 group strain condition name
## SlyA_control -19.12416 -17.395811 SlyA:control SlyA control SlyA_control
## SlyA_H2O2 26.21547 -3.552466 SlyA:H2O2 SlyA H2O2 SlyA_H2O2
## WT_control -28.90744 13.732492 WT:control WT control WT_control
## WT_H2O2 21.81613 7.215784 WT:H2O2 WT H2O2 WT_H2O2percentVar <- round(100 * attr(pcadata, "percentVar"))
library("ggplot2")
ggplot(pcadata, aes(PC1, PC2, color=condition, shape=strain)) + geom_point(size=3) +
xlab(paste0("PC1: ",percentVar[1],"% variance")) +
ylab(paste0("PC2: ",percentVar[2],"% variance"))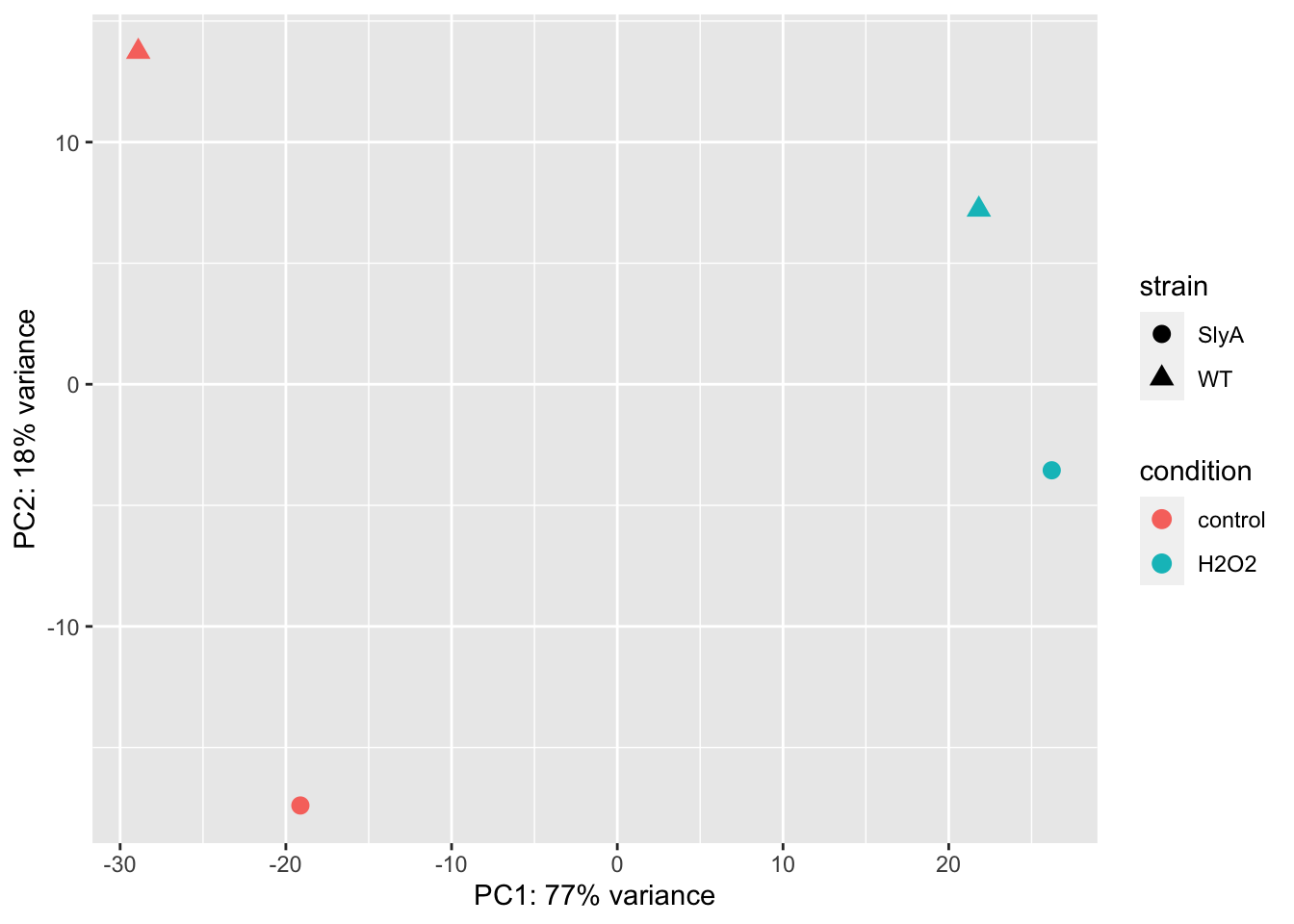
14.4.3.5 Análisis de expresión direfencial de genes
En DESeq2 el análisis difernecial es muy simple. Una vez creado el objeto DESeq2 con su diseño experimental, simplemente ejecutamos la función DESeq y obtenemos nuestro resultado.
## using pre-existing size factors## estimating dispersions## gene-wise dispersion estimates## mean-dispersion relationship## final dispersion estimates## fitting model and testingCon nuestro análisis almacenado en el objeto dds podemos construir nuestra tabla de resultados.
## log2 fold change (MLE): condition H2O2 vs control
## Wald test p-value: condition H2O2 vs control
## DataFrame with 4373 rows and 6 columns
## baseMean log2FoldChange lfcSE stat pvalue padj
## <numeric> <numeric> <numeric> <numeric> <numeric> <numeric>
## STy_00001 155.4201 0.0262375 0.602762 0.0435287 0.965280059 0.98381043
## STy_00002 5.4645 2.3912608 1.887756 1.2667212 0.205254987 NA
## STy_00003 105.2573 2.2846865 0.807069 2.8308426 0.004642556 0.02666813
## STy_00004 1110.7030 -1.8527954 0.501416 -3.6951278 0.000219776 0.00227855
## STy_00005 134.2055 1.7567197 0.656803 2.6746523 0.007480678 0.03828626
## ... ... ... ... ... ... ...
## STy_04504 1553.84391 0.3067794 0.496807 0.6175023 0.536903 0.728915
## STy_04505 1.95652 1.8272908 2.826115 0.6465733 0.517908 NA
## STy_04506 17.23692 0.6851926 1.090408 0.6283821 0.529754 NA
## STy_04507 10.27732 0.6497026 1.258901 0.5160871 0.605794 NA
## STy_04509 27.69201 0.0384563 0.858653 0.0447867 0.964277 0.983543Ahora el nuevo objeto res contiene los resultados de nuestro análisis. Podemos obtener una descripción más detallada con respecto a qué significan estos resultados:
## DataFrame with 6 rows and 2 columns
## type description
## <character> <character>
## baseMean intermediate mean of normalized counts for all samples
## log2FoldChange results log2 fold change (MLE): condition H2O2 vs control
## lfcSE results standard error: condition H2O2 vs control
## stat results Wald statistic: condition H2O2 vs control
## pvalue results Wald test p-value: condition H2O2 vs control
## padj results BH adjusted p-values- ¿Qué significan las columnas log2FoldChange y padj? ¿Qué significa un log2FoldChange de 5?
[?]Para obtener un resumen de cómo resultó el test de expresión diferencial, simplemente ejecutamos el siguiente comando:
##
## out of 4373 with nonzero total read count
## adjusted p-value < 0.1
## LFC > 0 (up) : 495, 11%
## LFC < 0 (down) : 581, 13%
## outliers [1] : 0, 0%
## low counts [2] : 340, 7.8%
## (mean count < 18)
## [1] see 'cooksCutoff' argument of ?results
## [2] see 'independentFiltering' argument of ?resultsVemos que a un nivel de falsos positivos (FDR) considerable del 10% obtenemos muchos genes sobre y sub expresados. Una manera de disminuir la cantidad de falsos positivos es siendo más estricto con el FDR además de filtrar por log2FoldChange.
##
## FALSE TRUE
## 2925 854##
## FALSE TRUE
## 3290 235No necesariamente nos intersa solo la comparación de acuerdo a la columna condition, es decir, “H2O2 vs. control”. También podríamos evaluar cambios en la expresión génica entre la cepa wild-type y la cepa mutante (SlyA). Para esto usamos nuevamente la función results pero esta vez con el argumento contrast.
## log2 fold change (MLE): strain SlyA vs WT
## Wald test p-value: strain SlyA vs WT
## DataFrame with 4373 rows and 6 columns
## baseMean log2FoldChange lfcSE stat pvalue padj
## <numeric> <numeric> <numeric> <numeric> <numeric> <numeric>
## STy_00001 155.4201 1.137072 0.602792 1.886343 0.0592487 0.568336
## STy_00002 5.4645 -1.963630 1.856030 -1.057973 0.2900677 NA
## STy_00003 105.2573 0.765590 0.806290 0.949522 0.3423552 0.958650
## STy_00004 1110.7030 1.014867 0.501413 2.024014 0.0429687 0.480699
## STy_00005 134.2055 0.759531 0.656336 1.157229 0.2471788 0.911939
## ... ... ... ... ... ... ...
## STy_04504 1553.84391 0.0715024 0.496806 0.143924 0.885560 0.995124
## STy_04505 1.95652 1.2070400 2.802646 0.430679 0.666702 NA
## STy_04506 17.23692 0.6523967 1.089189 0.598975 0.549190 NA
## STy_04507 10.27732 1.0897181 1.257631 0.866484 0.386225 NA
## STy_04509 27.69201 -0.4330956 0.858653 -0.504390 0.613988 NAEl primer término en contrast corresponde a la columna de la tabla de información de la muestra que se quiere usar para la comparación, mientras que el segundo y tercer término corresponde al númerador y denominador de la comparación, respectivamente.
En biología de alto rendimiento ( high-throughput biology ) tenemos que tener en cuenta que al hacer tantos tests estadísticos por azar podemos obtener falsos positivos. Debemos tener cuidado en no usar los valores de p-value sirectamente, sino que corregir por multiple testing. ¿Qué pasaría si simplemente establecemos el threshold de p-value a 0.05? Hay 1314 genes con un p-value menosr a 0.05 de un total de 4375 genes.
## [1] 1314## [1] 4373Ahora, supongamos por un momento que la hipótesis nula se cumple para todos los genes: ningún gen es afectado por el tratamiento con H2O2. Entonces, por definición de p-value, esperamos que el 5% de los genes tengan un p-value menor a 0.05. Esto serían 219 genes (4375*0.05). Si consideramos la lista de genes con un p-value menor a 0.05 que recién calculamos, 1314, entonces 219/1314 = 16.7 % de falsos positivos. Esto es sin duda inaceptable. Lo que normalmente se hace es ajustar el p-value con algún método estadístico como Benjamini-Hochberg y de esta manera controlar el número de falsos positivos. Si consideramos 10% de falsos positivos aceptable, entonces tenemos este número de genes diferencialmente expresados:
## [1] 1076- ¿Cuántos genes diferencialmente expresados habría si aceptamos un 1% de falsos positivos?
[?]Estos son los genes con la sobre expresión o up regulation más fuerte.
# Apartamos los genes con FDR < 10%
resSig <- subset(res, padj < 0.1)
# Miramos la lista de genes, ordenados por el valor en la columna log2FoldChange de mayor a menor
head(resSig[ order(resSig$log2FoldChange, decreasing=TRUE), ])## log2 fold change (MLE): condition H2O2 vs control
## Wald test p-value: condition H2O2 vs control
## DataFrame with 6 rows and 6 columns
## baseMean log2FoldChange lfcSE stat pvalue padj
## <numeric> <numeric> <numeric> <numeric> <numeric> <numeric>
## STy_01656 3497.57 10.53754 0.738509 14.2687 3.43030e-46 4.61147e-43
## STy_01657 4075.75 10.20493 0.669124 15.2512 1.61692e-52 6.52104e-49
## STy_00588 2040.27 7.11922 0.539580 13.1940 9.50035e-40 7.66298e-37
## STy_00577 6120.43 7.08529 0.575923 12.3025 8.78223e-35 3.93542e-32
## STy_00589 5739.74 6.95139 0.511771 13.5830 5.05061e-42 5.09228e-39
## STy_03372 2770.27 6.70932 0.526616 12.7404 3.52443e-37 2.03058e-34Ahora, visualicemos el conjunto de los resultados de acuerdo a su efecto biológico (log2FoldChange) y su número medio de cuentas o reads.

Los genes cuya expresión es significativamente diferente aparecen marcados con rojo de acuerdo a un umbral arbitrario. En este caso es FDR < 0.1. Veamos otra vez un MA plot pero esta vez con nuestros resultados filtrados por log2FoldChange.

Otra forma de visualizar los resultados es usando un mapa de calor o heatmap. En el heatmap realizado anteriormente comparábamos muestras contra muestras. En este caso queremos comparar muestras contra los genes que muestran más cambios.
library("genefilter")
topVarGenes <- head(order(rowVars(assay(rld)),decreasing=TRUE),20)
mat <- assay(rld)[ topVarGenes, ]
mat <- mat - rowMeans(mat)
df <- as.data.frame(colData(rld)[,c("strain", "condition")])
pheatmap(mat, annotation_col=df)
- ¿Cómo se comportan los genes con mayor cambio? ¿Cuál es la variable dominante en la expresión de estos genes?
[?]