Redes de co-ocurrencia de microorganismos
ISME Latin America, Septiembre 2019
0.1 Acerca del curso
El tutorial a continuación fue creado especialmente para guiar el trabajo práctico del curso pre-congreso ISME Latin America 2019: Análisis de datos bioinformáticos para metagenomas y amplicones usando R. A realizarse el próximo 9 y 10 de septiembre en la Universidad Técnica Federico Santa María, Valparaíso, Chile.
 Ir a la página de inicio del curso
Ir a la página de inicio del curso
El curso cuenta con 6 unidades. Ahora, usted se encuentra en la unidad: Redes de co-ocurrencia de microorganismos 
Otras unidades del curso son:
Introducción a R: Manipulación de datos y visualización
Análisis de secuencias de 16S con DADA2
Introducción a phyloseq y a análisis de diversidad
Búsqueda de genes de interés en datos de metagenómica shotgun
Visualización y curación de genomas ensamblados desde metagenomas (MAGs)




Profesor: Dr. Eduardo Castro-Nallar (eduardo.castro@unab.cl)
- Ayudantes:
- Dr. Florence Gutzwiller (florence.gutzwiller@gmail.com)
- M.Sc. Katterinne N. Mendez (mendez.katterinne@gmail.com)
Centro de Bioinformática y Biología Integrativa | Universidad Andrés Bello

1 Configurar sesión de R
Para trabajar en R, hay tres primeros pasos que debemos seguir: (1) cargar a los paquetes necesarios a la sesión actual, (2) configurar el directorio de trabajo, (3) e importar datos de entrada a la sesión actual.
En esta sección del tutorial, vamos a utilizar 15 paquetes de R en total:
Los siguientes 3 scripts te mostrarán una manera eficiente de instalar y cargar la lista de paquetes según su repositorio de origen, ya que cada repositorio tiene su propia función para instalar sus paquetes.
- Primero, enlistar los paquetes necesarios en diferentes vectores dependiendo de su repositorio de origen.
# Definir paquetes
# Repositorio CRAN
cran_packages <- c("bookdown", "knitr", "devtools", "igraph", "qgraph", "RColorBrewer", "tidyverse", "network", "sna", "grid", "gridExtra")
# Repositorio Bioconductor
bioc_packages <- c("phyloseq", "microbiome", "genefilter")
# Repositorio GitHub
git_source <- c("zdk123/SpiecEasi", "hallucigenia-sparsa/seqtime", "briatte/ggnet") # fuente/nombre del paquete
git_packages <- c("SpiecEasi", "seqtime", "ggnet") # nombre del paquete- Segundo, instalar los paquetes definidos arriba usando la función correspondiente a cada repositorio.
# Instalar paquetes CRAN
.inst <- cran_packages %in% installed.packages()
if(any(!.inst)) {
install.packages(cran_packages[!.inst])
}
# Intalar paquetes BioConductor
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
.inst <- bioc_packages %in% installed.packages()
if(any(!.inst)) {
BiocManager::install(bioc_packages[!.inst])
}
# Instalar paquetes GitHub
.inst <- git_source %in% installed.packages()
if(any(!.inst)) {
devtools::install_github(git_source[!.inst])
}- Tercero, cargar los paquetes requeridos a la sesión actual de R.
# Cargar paquetes
sapply(c(cran_packages, bioc_packages, git_packages), require, character.only = TRUE)El paso de instalación de paquetes es necesario solamente una vez. Excepto si se quiere actualizar la versión del paquete, o bien, R ha sido desinstalado e instalado nuevamente o actualizado su versión.
Si ya tienes los paquetes instalados en tu computadora, sólo necesitas enlistar (1er script) y cargar (3er script) los paquetes. También, puedes cargarlos a la sesión actual de R usando la función library(), así:
# Cargar paquetes
library(phyloseq)
library(microbiome)
library(genefilter)
library(SpiecEasi)
library(seqtime)
library(igraph)
library(qgraph)
library(ggnet)
library(RColorBrewer)
library(tidyverse)
library(grid)
library(gridExtra)
library(network)
library(sna)
library(ggplot2)2 Datos de estudio
El último paso antes de comenzar, es cargar y pre-procesar los datos de entrada. Para hacer análisis de redes de co-ocurrencia de microorganismos, necesitamos el perfil taxonómico y de abundancia de cada microorganismo detectado en todas las muestras que incluye el estudio. Dependiendo si los datos fueron generados por secuenciación de DNA total o de amplicones (shotgun sequencing o amplicon sequencing, respectivamente), hay varios caminos hasta obtener el análisis de composición microbiana.
A continuación, paso a describir brévemente el origen y procesamiento de los datos a utilizar en esta sección del curso.
En este tutorial, vamos a trabajar 69 metagenomas producto de secuenciación de tipo shotgun de muestras de agua colectadas en el fiordo Comau ubicado en las costas de la Patagonia norte chilena, en la Región de Los Lagos.
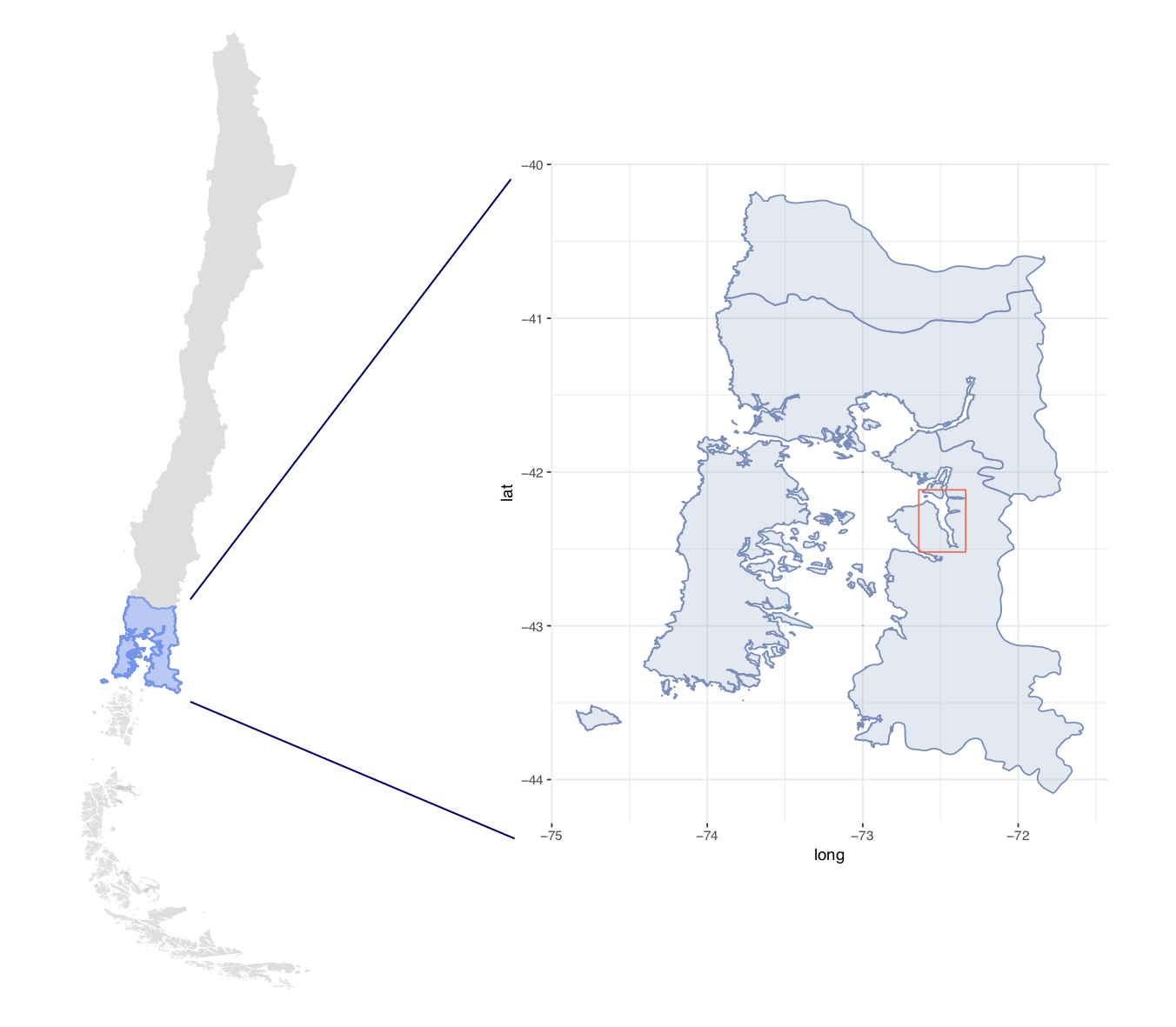
Localización geográfica del fiordo Comau

Fiordo Comau: vista desde camino al Cerro El Tambor
Fuente: flickr.com/photos/lalo_pangue
En el fiordo Comau, las muestras fueron tomadas en 15 sitios diferentes, desde dos profundidades (5 y 20 metros), y en 5 fechas incluyendo los años 2016, 2017, y 2018.
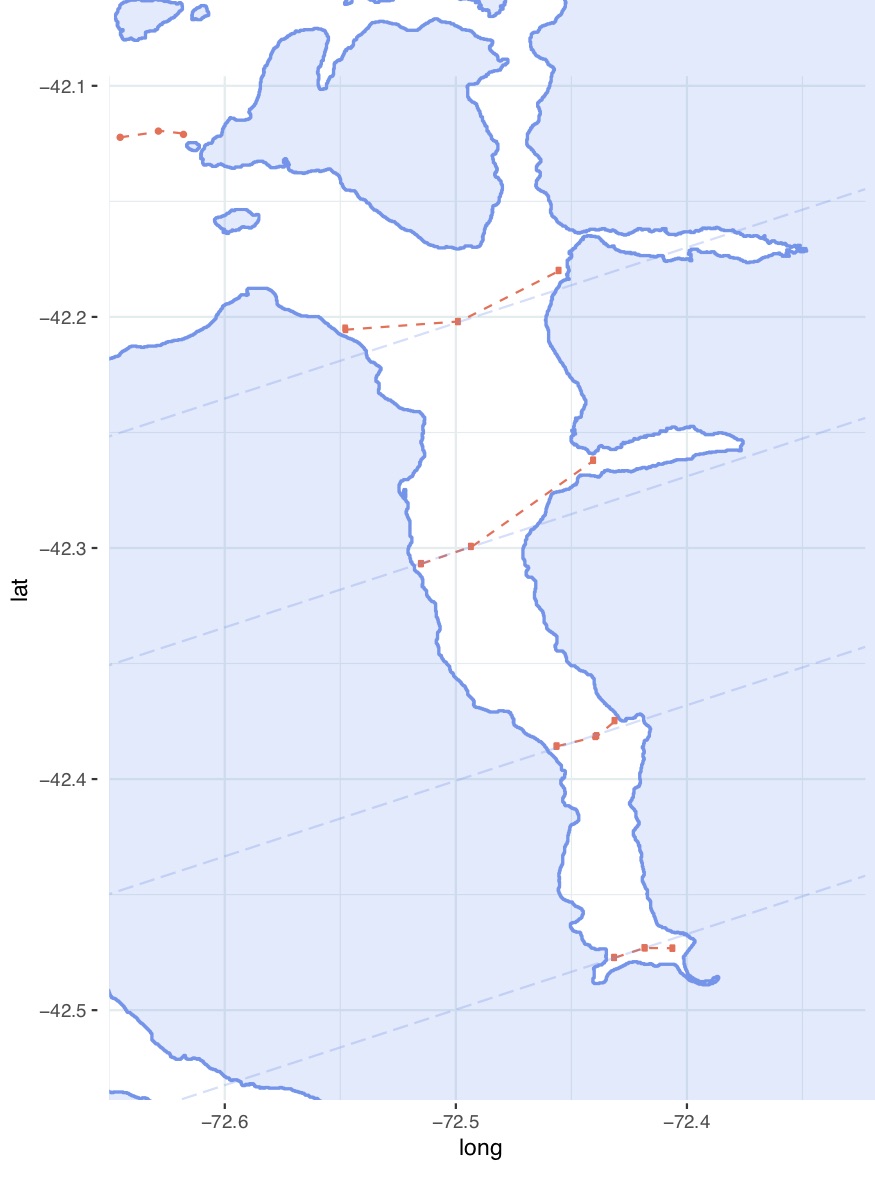
Sitios de toma de muestras en el fiordo Comau
Las muestras de ~25 L de agua fueron filtradas usando filtros Sterivex de 0.22 um para retener células procariontes. Desde los filtros se extrajo DNA total y se prepararon librerías paired-end siguiendo la guía de preparación de muestras TruSeq de Illumina. Finalmente, las 69 librerías de tipo paired-end fueron enviadas a Macrogen para ser secuenciadas en la plataforma Illumina HiSeq 3000.
Las reads o raw data junto con la metadata (i.e., datos ambientales tomados junto con las muestras) de los metagenomas marinos colectados en el fiordo Comau se encuentran disponibles en la base de datos NCBI bajo los BioProject PRJNA359936 y PRJNA517740.
Las secuencias metagenómicas de las 69 muestras fueron pre-procesadas para eliminar adaptadores, secuencias/bases de baja calidad (i.e., quality score < 20), y secuencias cortas (i.e., largo < 50 pb), usando PRINSEQ. En total, 2.7 mil millones de secuencias (después del control de calidad) fueron utilizadas para los análisis de composición microbiana.
Para estimar la composición microbiana de cada muestra, primero las reads fueron clasificadas usando Centrifuge en contra de una base de datos personalizada que incluye la base de datos GTDB (Genome Taxonomy Database), los genomas ensamblados desde metagenomas (MAGs) del Tara Oceans (global oceans expedition), y los genomas ensamblados desde metagenomas del mismo fiordo Comau.
Para más información acerca de por qué utilizar una base de datos personalizada para clasificar las reads metagenómicas y como esta estrategía puede aumentar significativamente el porcentaje de secuencias clasificadas, y en consecuencia, su asignación taxonómica, especialmente en el caso de metagenomas ambientales:
Guillaume Méric, Ryan R. Wick, Stephen C. Watts, Kathryn E. Holt, Michael Inouye. 2019. Correcting index databases improves metagenomic studies. bioRxiv, doi.org/10.1101/712166.
Finalmente, a partir de la clasificación de las secuencias de cada uno de los 69 metagenomas del fiordo Comau, se generó el objeto phyloseq que contiene la información de abundancia y perfil taxonómico de cada microorganismo detectado en cada muestra, junto con la metadata (i.e., datos ambientales) de cada una. Desde este punto en adelante, trabajamos el análisis de composición microbiana y de redes de co-ocurrencia en R.
2.1 Datos de entrada
Para construir una red de co-ocurrencia de microorganismos, como mínimo necesitamos una tabla de abundancia de todas las taxas identificadas en cada una de las muestras (también conocida como OTU table), ya que esta se utiliza para calcular la posible relación entre diferentes microorganismos o taxas de acuerdo a su abundancia a través de las muestras. Por lo tanto, el dato de entrada o input para construir una red de co-ocurrencia puede ser tanto una OTU table o un objeto phyloseq.
En este tutorial, el dato de entrada o input será el objeto phyloseq: comau69_3
Antes de cargar el objeto phyloseq
comau69_3, DESCARGA EL INPUTcomau69_3.RDSAQUÍCarga el objeto phyloseq a tu sesión de R actual, así:
# Cargar objeto phyloseq
comau69_3 <- readRDS(file = "comau69_3.RDS")
comau69_3 # overview## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 650 taxa and 69 samples ]
## sample_data() Sample Data: [ 69 samples by 23 sample variables ]
## tax_table() Taxonomy Table: [ 650 taxa by 7 taxonomic ranks ] IMPORTANTE: El objeto phyloseq
IMPORTANTE: El objeto phyloseq comau69_3 ya se encuentra procesado y esta listo para inferir la red de co-ocurrencia de microorganismos del fiordo Comau.
 ¿A qué nos referimos con procesado? Antes de inferir redes de co-ocurrencia, se recomienda aglomerar la taxonomía por el best hit y filtrar por prevalencia (ver abajo en “Pre-procesamiento” para más detalle). Con el fin de reducir la heterogeneidad de las muestras, lo que según estudios basados en datos simulados, mejora la precisión de la red.
¿A qué nos referimos con procesado? Antes de inferir redes de co-ocurrencia, se recomienda aglomerar la taxonomía por el best hit y filtrar por prevalencia (ver abajo en “Pre-procesamiento” para más detalle). Con el fin de reducir la heterogeneidad de las muestras, lo que según estudios basados en datos simulados, mejora la precisión de la red.
Si no estás interesado en conocer los pasos de pre-procesamiento (i.e., aglomerar y filtrar) de los datos de estudio hasta obtener el objeto comau69_3, puedes saltar los pasos a continuación y seguir con el cálculo de la red en la sección 3.
De lo contrario, puedes cargar el objeto phyloseq inicial comau69_1 y seguir los pasos de pre-procesamiento.
Antes de cargar el objeto phyloseq
comau69_1, DESCARGA EL INPUTcomau69_1.RDSAQUÍCarga el objeto phyloseq a tu sesión de R actual, así:
# Leer objeto phyloseq
comau69_1 <- readRDS(file = "comau69_1.RDS")
comau69_1 # overview## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 25361 taxa and 69 samples ]
## sample_data() Sample Data: [ 69 samples by 23 sample variables ]
## tax_table() Taxonomy Table: [ 25361 taxa by 7 taxonomic ranks ]2.1.1 Pre-procesamiento
2.1.1.1 Aglomerar por best hit
Aglomerar por el best hit se refiere a que el proceso de aglomeración se hará según el linaje completo y no por un rango taxonómico en particular.
Para ello, primero utilizaremos la función format_to_besthit() del paquete microbiomeutilities sobre el objeto phyloseq comau69_1, para agregar a la tabla de taxonomía el último rango alcanzado en la clasificación taxonómica de cada taxa (best hit). Seguidamente, usamos la función tax_glom del paquete phyloseq para aglomerar según la octava columna, que contiene el último rango taxonómico alcanzado.
- Así:
# Agregar la mejor taxonomía en el objeto phyloseq
comau69_2 <- microbiomeutilities::format_to_besthit(comau69_1)
comau69_2 #overview
# La función 'format_to_besthit()' hace varias modificaciones en el objeto phyloseq, que por conveniencia vamos a revertir:
# Nombre de las taxas o filas de la tabla de taxonomía (tax table)
rownames(comau69_2@tax_table@.Data) <- rownames(comau69_1@tax_table@.Data)
# Nombre de las taxas o filas de la tabla de abundancia (otu table)
rownames(comau69_2@otu_table@.Data) <- rownames(comau69_1@otu_table@.Data)
# Remover la columna extra creada por 'format_to_besthit()' en la tax table
tax_table(comau69_2) <- tax_table(comau69_2)[,1:7]
# Reemplazar el título de la columna "Domain" por "Kingdom" en la tax table
colnames(comau69_2@tax_table@.Data) <- c("Kingdom", "Phylum", "Class", "Order", "Family", "Genus", "Species")
# Mira como quedó la tabla de taxonomía en el objeto phyloseq
View(comau69_2@tax_table@.Data)
# Aglomerar por el best hit, usando el último rango taxonómico: "Species"
comau69_2 <- tax_glom(comau69_2, "Species", NArm = FALSE)
comau69_2 #overview Ambos pasos, calcular el best hit y aglomerar, pueden tomar varios minutos o incluso unas pocas horas. Para ahorrar una espera innecesaria y seguir avanzando en el tutorial, descarga el objeto phyloseq comau69_2 AQUÍ
- Carga el objeto phyloseq a tu sesión de R actual, así:
# Cargar objeto phyloseq
comau69_2 <- readRDS(file = "comau69_2.RDS")
comau69_2 #overview## phyloseq-class experiment-level object
## otu_table() OTU Table: [ 24218 taxa and 69 samples ]
## sample_data() Sample Data: [ 69 samples by 23 sample variables ]
## tax_table() Taxonomy Table: [ 24218 taxa by 7 taxonomic ranks ]2.1.1.2 Filtrar
El filtro por prevalencia reduce el número de taxas eliminando aquellas que tengan 0 cuentas (read count) en un porcentaje del número total de muestras definido por el valor de prevalencia.
Como se explica en el review de Lisa Rottjers y Karoline Faust: From hairballs to hypotheses–biological insights from microbial networks. No somos capaces de discriminar si los valores igual a cero en la tabla de abundancia (otu table) representan la ausencia de la taxa o un muestreo insuficiente.
¿Qué valor de prevalencia usar? No hay una guía o valor definido aún, pero en el review mencionado arriba de Lisa Rottjers y Karoline Faust, recomiendan definir el valor de prevalencia apuntando a un equilibrio entre: evitar asociaciones erradas y conservar potenciales especies importantes. Por lo tanto, depende de los datos de estudio y/o de la pregunta biológica.
Por ejemplo, en el caso de los datos que trabajamos en este tutorial: Estamos interesados en observar las taxas de baja abundancia (rare taxa), ya que creemos podrían tener un rol importante en la comunidad microbiana del fiordo. Entonces, previamente experimentamos haciendo filtros por prevalencia y por abundancia relativa. En seguida descubrimos que al filtrar por abundancia relativa, todas las taxas que pasaron el criteri de filtro presentaban una alta prevalencia. Finalmente, elegimos este criterio de filtro porque nos permite conservar las taxas de baja abundancia manteniendo altos los valores de prevalencia.
- Filtrar taxas por abundancia relativa:
- Primero, usamos la función
transform_sample_counts()para calcular los valores de abundancia relativa (número de reads clasificados dividido por el número total de reads). - Segundo, usamos la función
kOverA()del paquete genefilter para definir el criterio de filtro:
conservar aquellas taxas con una abundancia relativa mayor a 0.05% en al menos 1 de las 69 muestras - Tercero, usamos la función
prune_taxa()para filtrar el objeto phyloseq.
- Primero, usamos la función
# Transformar los valores de cuentas (otu table) a valores de abundancia relativa en el objeto phyloseq
comau69_2ra <- transform_sample_counts(comau69_2, function(x) x / sum(x))
# Definir criterio de filtro usando la función 'kOverA(k, A)'
# k = número de muestras; A = vaor de abundancia
# k = 1; A = 0.0005
flist <- filterfun(kOverA(1, 0.0005))
# Filtrar taxas
# Crear objeto que clasifica como TRUE a aquellas taxas que pasaron el criterio de filtro
comau69_2raf <- filter_taxa(comau69_2ra, flist)
# Filtrar el objeto phyloseq
comau69_3 <- prune_taxa(comau69_2raf, comau69_2)
comau69_3 #overview# Mira la tabla de abundancia
View(comau69_3@otu_table@.Data)| AUG2018_L1S1D05 | AUG2018_L1S1D20 | AUG2018_L1S2D05 | AUG2018_L1S2D20 | AUG2018_L1S3D05 | AUG2018_L1S3D20 | AUG2018_L3S1D05 | AUG2018_L3S1D20 | AUG2018_L3S2D05 | AUG2018_L3S2D20 | AUG2018_L3S3D05 | AUG2018_L3S3D20 | AUG2018_L4S1D05 | AUG2018_L4S1D20 | AUG2018_L4S2D05 | AUG2018_L4S2D20 | AUG2018_L4S3D05 | AUG2018_L4S3D20 | AUG2018_L5S1D05 | AUG2018_L5S1D20 | AUG2018_L5S2D05 | AUG2018_L5S2D20 | AUG2018_L5S3D05 | AUG2018_L5S3D20 | BacS1 | BacS2 | BacS3 | DEC2017_L1S1D05 | DEC2017_L1S1D20 | DEC2017_L1S2D05 | DEC2017_L1S3D05 | DEC2017_L1S3D20 | DEC2017_L3S1D05 | DEC2017_L3S1D20 | DEC2017_L3S3D05 | DEC2017_L3S3D20 | DEC2017_L4S1D05 | DEC2017_L4S1D20 | DEC2017_L4S2D05 | DEC2017_L4S2D20 | DEC2017_L4S3D05 | DEC2017_L4S3D20 | DEC2017_L5S1D05 | DEC2017_L5S1D20 | DEC2017_L5S2D05 | DEC2017_L5S2D20 | DEC2017_L5S3D05 | DEC2017_L5S3D20 | JAN2018_L3S2D05 | JAN2018_L3S2D20 | JUN2017_L1S1D05 | JUN2017_L1S2D05 | JUN2017_L1S2D20 | JUN2017_L1S3D05 | JUN2017_L1S3D20 | JUN2017_L3S1D20 | JUN2017_L3S2D05 | JUN2017_L3S2D20 | JUN2017_L4S1D05 | JUN2017_L4S1D20 | JUN2017_L4S2D05 | JUN2017_L4S2D20 | JUN2017_L4S3D05 | JUN2017_L4S3D20 | JUN2017_L5S1D05 | JUN2017_L5S1D20 | JUN2017_L5S2D05 | JUN2017_L5S2D20 | JUN2017_L5S3D05 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TI_27683 | 2132914 | 446684 | 1828370 | 734858 | 2002908 | 481932 | 344432 | 34090 | 593656 | 30588 | 754466 | 39859 | 657003 | 92631 | 1108959 | 517165 | 1411538 | 228912 | 392922 | 431466 | 1752318 | 569986 | 1026325 | 249799 | 62043 | 55477 | 169719 | 859509 | 899814 | 1488533 | 2356204 | 1842909 | 1686912 | 719965 | 802211 | 1174158 | 2064040 | 668016 | 561522 | 784329 | 217988 | 262094 | 1300809 | 557228 | 770428 | 432943 | 591262 | 160154 | 357727 | 38925 | 104981 | 104914 | 24947 | 88872 | 46080 | 3126 | 216776 | 6650 | 145223 | 26183 | 116443 | 31726 | 74114 | 20554 | 71942 | 22783 | 138095 | 32185 | 58110 |
| TI_39035 | 977 | 10787 | 922 | 4144 | 1227 | 3579 | 256 | 11418 | 409 | 10795 | 358 | 16686 | 764 | 1333 | 1422 | 7124 | 1742 | 3171 | 418 | 2214 | 1134 | 7171 | 2766 | 6847 | 97 | 102 | 108 | 19951 | 1664 | 1191 | 3103 | 3134 | 1688 | 1522 | 1506 | 290 | 537 | 3020 | 8097 | 2110 | 6464 | 2558 | 633 | 1237 | 409 | 6408 | 560 | 338 | 1197 | 22969 | 20645 | 19193 | 14628 | 12961 | 14597 | 8239 | 7196 | 11890 | 27096 | 12297 | 25549 | 13062 | 17103 | 13099 | 21582 | 13793 | 23239 | 16852 | 20818 |
| TI_27681 | 129 | 924 | 119 | 399 | 225 | 398 | 66 | 861 | 124 | 924 | 93 | 1379 | 131 | 144 | 187 | 691 | 213 | 340 | 59 | 268 | 201 | 836 | 306 | 776 | 26 | 38 | 43 | 2079 | 337 | 415 | 413 | 407 | 449 | 291 | 380 | 103 | 116 | 439 | 969 | 385 | 715 | 536 | 150 | 251 | 158 | 776 | 215 | 88 | 218 | 1605 | 2367 | 1940 | 1902 | 2067 | 1721 | 1541 | 769 | 1441 | 3090 | 1711 | 2588 | 1880 | 1902 | 1937 | 2941 | 1850 | 2734 | 4487 | 3100 |
| TI_13593 | 1308850 | 3365486 | 1022844 | 3040117 | 1482830 | 1519207 | 181828 | 86186 | 1081413 | 161198 | 163905 | 131914 | 1481758 | 355686 | 680570 | 1069106 | 628357 | 534329 | 563609 | 443793 | 382266 | 1069965 | 600898 | 492585 | 13185 | 7373 | 3118 | 2683197 | 7798839 | 1319276 | 813638 | 5579080 | 4740967 | 4354461 | 2895557 | 57235 | 22516 | 2663175 | 1013002 | 1360649 | 662193 | 1023236 | 1110980 | 1726956 | 325612 | 870570 | 1062262 | 612162 | 2105090 | 215064 | 84480 | 83334 | 43519 | 63473 | 53791 | 10913 | 31560 | 17519 | 144717 | 48971 | 113167 | 49273 | 85574 | 40743 | 91391 | 43532 | 150608 | 70097 | 91030 |
| TI_13597 | 66037 | 162935 | 51765 | 145039 | 76116 | 77887 | 10286 | 6393 | 49574 | 11305 | 10898 | 9940 | 63831 | 16313 | 37926 | 55858 | 34506 | 25550 | 25168 | 23760 | 25269 | 54498 | 29336 | 24772 | 6891 | 5184 | 5519 | 132432 | 332103 | 126603 | 52765 | 246777 | 235951 | 192132 | 150925 | 11465 | 11240 | 124679 | 56481 | 71910 | 35922 | 55909 | 60453 | 75991 | 22596 | 48159 | 73717 | 35187 | 97733 | 18158 | 9266 | 8140 | 5917 | 6710 | 6161 | 3041 | 3739 | 3478 | 15342 | 6228 | 11461 | 5866 | 8997 | 5711 | 10770 | 5313 | 13402 | 10428 | 10334 |
| TI_20829 | 393029 | 172057 | 360412 | 303746 | 420787 | 188589 | 75319 | 25570 | 156945 | 18705 | 104920 | 24515 | 167470 | 38208 | 326174 | 258648 | 362785 | 89202 | 72003 | 98090 | 249539 | 142447 | 160137 | 51917 | 6799 | 8382 | 19167 | 180635 | 458225 | 478760 | 271704 | 1150808 | 891532 | 470387 | 362761 | 40185 | 52298 | 435196 | 98043 | 478134 | 64956 | 170954 | 293849 | 230265 | 151000 | 146600 | 218217 | 96803 | 153469 | 92346 | 19579 | 16178 | 13214 | 13754 | 13417 | 8175 | 36433 | 13855 | 22660 | 8457 | 24152 | 9814 | 14531 | 10117 | 19200 | 9753 | 23875 | 12439 | 19371 |
| TI_20846 | 32501 | 77289 | 28404 | 89361 | 40890 | 74012 | 9592 | 18632 | 10981 | 12355 | 10560 | 23748 | 49911 | 31150 | 68516 | 179372 | 98469 | 57929 | 15858 | 38460 | 78282 | 67337 | 44081 | 40673 | 431 | 358 | 1111 | 17727 | 10963 | 6605 | 4741 | 23519 | 9486 | 20837 | 17086 | 2556 | 2695 | 65654 | 9647 | 95531 | 5151 | 95651 | 3756 | 19587 | 4146 | 29780 | 3654 | 4469 | 15533 | 78958 | 25553 | 21644 | 19047 | 19444 | 20417 | 11679 | 42075 | 21320 | 22757 | 10585 | 26599 | 12592 | 18414 | 15669 | 24264 | 12581 | 22955 | 18478 | 25749 |
| TI_20835 | 2434 | 3772 | 2009 | 4265 | 2720 | 3480 | 905 | 1102 | 789 | 677 | 1165 | 1315 | 2457 | 1464 | 3260 | 8527 | 4818 | 2763 | 806 | 1825 | 4258 | 3308 | 2530 | 1856 | 71 | 66 | 180 | 1595 | 1358 | 1132 | 1380 | 2329 | 1567 | 1689 | 1636 | 665 | 1162 | 3909 | 845 | 5205 | 405 | 4688 | 792 | 1445 | 511 | 1937 | 501 | 350 | 1221 | 3855 | 1213 | 1008 | 825 | 888 | 983 | 522 | 2027 | 1043 | 1168 | 444 | 1235 | 574 | 885 | 731 | 1098 | 616 | 1127 | 721 | 1169 |
| TI_38896 | 13288 | 37930 | 11716 | 18187 | 15047 | 18470 | 2058 | 36028 | 3334 | 24654 | 5109 | 36952 | 4299 | 4942 | 8637 | 23567 | 11472 | 12168 | 2511 | 6054 | 11161 | 16002 | 9700 | 12136 | 682 | 720 | 687 | 103719 | 8010 | 11988 | 20221 | 20594 | 14160 | 7664 | 8665 | 2679 | 3638 | 16592 | 41688 | 11112 | 29969 | 14304 | 5103 | 6285 | 3124 | 19591 | 4680 | 2180 | 5879 | 86420 | 24916 | 20712 | 19409 | 14991 | 19921 | 12390 | 14697 | 21997 | 46691 | 17502 | 27861 | 18391 | 21256 | 17486 | 25921 | 17560 | 30822 | 30402 | 28667 |
| TI_20841 | 426 | 6518 | 351 | 2822 | 626 | 3182 | 84 | 3744 | 81 | 3856 | 84 | 7114 | 600 | 1022 | 987 | 3706 | 990 | 2376 | 374 | 2429 | 662 | 6623 | 1921 | 5996 | 287 | 364 | 137 | 55400 | 439 | 646 | 7131 | 3261 | 815 | 793 | 709 | 69 | 94 | 2968 | 20731 | 959 | 18700 | 2160 | 386 | 742 | 152 | 12453 | 547 | 337 | 897 | 13131 | 23237 | 18677 | 13156 | 13275 | 16616 | 6993 | 8419 | 14186 | 24178 | 16217 | 24395 | 14474 | 19045 | 12208 | 25795 | 13718 | 27605 | 21907 | 24301 |
- Puedes descargar el objeto phyloseq
comau69_3AQUÍ y cargarlo a la sesión de R actual.
# Leer objeto phyloseq
comau69_3 <- readRDS(file = "comau69_3.RDS")
comau69_3 # overviewphyloseq-class experiment-level object
otu_table() OTU Table: [ 650 taxa and 69 samples ]
sample_data() Sample Data: [ 69 samples by 23 sample variables ]
tax_table() Taxonomy Table: [ 650 taxa by 7 taxonomic ranks ]2.2 Perfil taxonómico
Puede ser muy útil tener una tabla de datos disponible para consultar durante el análisis.
- Vamos a usar la función
psmeltpara tabular la información contenida en el objeto phyloseqcomau69_3
# Extraer y tabular los datos en comau69_3
taxotutable <- phyloseq::psmelt(comau69_3)
# Editar tabla
colnames(taxotutable) <- c("TaxID", "Sample", "Abundance",
"sample_ID", "sample_date", "sample_name", "line", "site", "line_site", "depth_m", "latitude", "longitude", "lat_lon", "temperature_C", "light_lux", "pH", "salinity_PSU", "chlorophyll_A_ugL", "C_total_ug", "N_total_ug", "C_N", "nitrate_nitrite_uM", "nitrate_uM", "nitrite_uM", "phosphate_uM", "silicate_uM",
"Kingdom", "Phylum", "Class", "Order", "Family", "Genus", "Species")
# Exportar/guardar tabla
write.table(taxotutable, file = "SummarizedTaxAssigments_comau69_3.tsv", sep = "\t", quote = FALSE)- También, puedes descargar la tabla
SummarizedTaxAssigments_comau69_3.tsvAQUÍ y cargarla a la sesión actual de R.
# Leer y guardar tabla en objeto 'taxotutable'
taxotutable <- read.table(file = "SummarizedTaxAssigments_comau69_3.tsv", sep = "\t", header = TRUE)- Usa la función
View()para visualizar la tabla de datostaxotutableen RStudio.
# Leer y guardar tabla en objeto 'taxotutable'
taxotutable <- read.table(file = "SummarizedTaxAssigments_comau69_3.tsv", sep = "\t", header = TRUE)| TaxID | Sample | Abundance | sample_ID | sample_date | sample_name | line | site | line_site | depth_m | latitude | longitude | lat_lon | temperature_C | light_lux | pH | salinity_PSU | chlorophyll_A_ugL | C_total_ug | N_total_ug | C_N | nitrate_nitrite_uM | nitrate_uM | nitrite_uM | phosphate_uM | silicate_uM | Kingdom | Phylum | Class | Order | Family | Genus | Species |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TI_13593 | DEC2017_L1S1D20 | 7798839 | DEC2017_L1S1D20 | DEC2017 | L1S1D20 | L1 | S1 | L1S1 | 20 | -42.47733 | -72.43112 | 42.47733 S 2.43112 W | 12.87 | 75.330 | 8.27 | 32.18 | 0.1455 | 55.2030 | 6.5863 | 8.3815 | 0.8000 | 0.8000 | 0.0000 | 0.8800 | 2.0000 | Bacteria | Proteobacteria | Alphaproteobacteria | Rhodobacterales | Rhodobacteraceae | Amylibacter_A | sp000153745 |
| TI_13593 | DEC2017_L1S3D20 | 5579080 | DEC2017_L1S3D20 | DEC2017 | L1S3D20 | L1 | S3 | L1S3 | 20 | -42.47310 | -72.40573 | 42.4731 S 2.40573 W | 13.48 | NA | 8.33 | 32.08 | 0.9275 | 58.2057 | 6.3085 | 9.2266 | 0.6000 | 0.6000 | 0.0000 | 0.6300 | 1.0000 | Bacteria | Proteobacteria | Alphaproteobacteria | Rhodobacterales | Rhodobacteraceae | Amylibacter_A | sp000153745 |
| TI_26133 | DEC2017_L1S1D20 | 4968562 | DEC2017_L1S1D20 | DEC2017 | L1S1D20 | L1 | S1 | L1S1 | 20 | -42.47733 | -72.43112 | 42.47733 S 2.43112 W | 12.87 | 75.330 | 8.27 | 32.18 | 0.1455 | 55.2030 | 6.5863 | 8.3815 | 0.8000 | 0.8000 | 0.0000 | 0.8800 | 2.0000 | Bacteria | Proteobacteria | Gammaproteobacteria | Pseudomonadales | Pseudohongiellaceae | OM182 | sp001438145 |
| TI_13593 | DEC2017_L3S1D05 | 4740967 | DEC2017_L3S1D05 | DEC2017 | L3S1D05 | L3 | S1 | L3S1 | 5 | -42.38636 | -72.45599 | 42.38636 S 2.45599 W | 15.02 | 403.625 | 8.54 | 32.72 | 0.3819 | 56.0702 | 6.4933 | 8.6351 | 0.0000 | 0.0000 | 0.0100 | 0.4500 | 1.0000 | Bacteria | Proteobacteria | Alphaproteobacteria | Rhodobacterales | Rhodobacteraceae | Amylibacter_A | sp000153745 |
| TI_39096 | AUG2018_L3S3D05 | 4557864 | AUG2018_L3S3D05 | AUG2018 | L3S3D05 | L3 | S3 | L3S3 | 5 | -42.37470 | -72.43074 | 42.3747 S 72.43074 W | 10.44 | 53.800 | 8.09 | 30.16 | NA | NA | NA | NA | 12.7048 | 12.5244 | 0.1804 | 1.2357 | 2.6795 | Bacteria | Proteobacteria | Alphaproteobacteria | Rhodobacterales | Rhodobacteraceae | Tateyamaria | g__Tateyamaria |
| TI_13593 | DEC2017_L3S1D20 | 4354461 | DEC2017_L3S1D20 | DEC2017 | L3S1D20 | L3 | S1 | L3S1 | 20 | -42.38636 | -72.45599 | 42.38636 S 2.45599 W | 12.83 | 43.050 | 8.04 | 34.06 | 0.2364 | 41.5871 | 2.4199 | 17.1856 | 0.9000 | 0.9000 | 0.0000 | 0.9200 | 0.0000 | Bacteria | Proteobacteria | Alphaproteobacteria | Rhodobacterales | Rhodobacteraceae | Amylibacter_A | sp000153745 |
| TI_26133 | DEC2017_L3S1D05 | 3706850 | DEC2017_L3S1D05 | DEC2017 | L3S1D05 | L3 | S1 | L3S1 | 5 | -42.38636 | -72.45599 | 42.38636 S 2.45599 W | 15.02 | 403.625 | 8.54 | 32.72 | 0.3819 | 56.0702 | 6.4933 | 8.6351 | 0.0000 | 0.0000 | 0.0100 | 0.4500 | 1.0000 | Bacteria | Proteobacteria | Gammaproteobacteria | Pseudomonadales | Pseudohongiellaceae | OM182 | sp001438145 |
| TI_27699 | AUG2018_L3S3D05 | 3466513 | AUG2018_L3S3D05 | AUG2018 | L3S3D05 | L3 | S3 | L3S3 | 5 | -42.37470 | -72.43074 | 42.3747 S 72.43074 W | 10.44 | 53.800 | 8.09 | 30.16 | NA | NA | NA | NA | 12.7048 | 12.5244 | 0.1804 | 1.2357 | 2.6795 | Bacteria | Proteobacteria | Alphaproteobacteria | Rhodobacterales | Rhodobacteraceae | Planktotalea | sp000156115 |
| TI_13593 | AUG2018_L1S1D20 | 3365486 | AUG2018_L1S1D20 | AUG2018 | L1S1D20 | L1 | S1 | L1S1 | 20 | -42.47733 | -72.43112 | 42.47733 S 72.43112 W | 10.88 | NA | 7.79 | 29.82 | NA | NA | NA | NA | 22.1702 | 21.7523 | 0.4179 | 2.2719 | 9.7044 | Bacteria | Proteobacteria | Alphaproteobacteria | Rhodobacterales | Rhodobacteraceae | Amylibacter_A | sp000153745 |
| TI_23044 | DEC2017_L3S3D20 | 3323790 | DEC2017_L3S3D20 | DEC2017 | L3S3D20 | L3 | S3 | L3S3 | 20 | -42.37470 | -72.43074 | 42.3747 S 2.43074 W | 12.62 | 0.000 | 8.38 | 34.69 | 0.4001 | 47.7105 | 4.1446 | 11.5115 | 0.9000 | 0.9000 | 0.0000 | 0.8900 | 0.0000 | Bacteria | Bacteroidota | Bacteroidia | Flavobacteriales | Flavobacteriaceae | MAG-120531 | sp000173115 |
3 Redes de co-ocurrencia microbiana
Las as redes de co-ocurrencia nos permiten estudiar la comunidad microbiana más allá de solamente su composición: detección de keystone species, entender mejor la dinámica de la comunidad microbiana, y analizar efecto de factores abióticos sobre la comunidad microbiana.
En este tutorial, vamos a calcular la red, conocer sus características 4, calcular estadística (degree, centrality, transitivity) 5, estructura de la red (detección y extracción de módulos/clusters) 6, y búsqueda de keystone species 7.
 Este tutorial se basa fuertemente en el trabajo de otros investigadores publicado en:
Este tutorial se basa fuertemente en el trabajo de otros investigadores publicado en:
Zachary Kurtz. github.com/zdk123/SpiecEasi
Mehdi Layeghifard, David M. Hwang, and David S. Guttman. 2018. Constructing and Analyzing Microbiome Networks in R. Microbiome Analysis, Methods and Protocols. doi.org/10.1007/978-1-4939-8728-3_16
Karoline Faust. Microbial association network construction tutorial
ggnet2: network visualization with ggplot2
ggnetwork: network geometries for ggplot2
Katherine Ognyanova. Network visualization with R
3.1 SPIEC-EASI
SPIEC-EASI (SParse InversE Covariance estimation for Ecological Association Inference) es un método estadístico para la inferencia de redes basado en la covariancia inversa entre las taxas según sus valores de abundancia a través de las muestras. Para conocer más acerca de SPIEC-EASI, revisa el siguiente artículo:
Zachary D. Kurtz, Christian L. Müller, Emily R. Miraldi, Dan R. Littman, Martin J. Blaser, Richard A. Bonneau. 2015. Sparse and Compositionally Robust Inference of Microbial Ecological Networks. PLoS Computational Biology. doi.org/10.1371/journal.pcbi.1004226
- Usamos la función
spiec.easi()para inferir la red a partir del objeto phyloseqcomau69_3y la guardamos en el objetose_mb
No necesitas normalizar la tabla de abundancia (otu table), por ejemplo, transformando los valores de cuentas a abundancia relativa. SPIEC-EASI se encarga de esto transformando los valores de cuentas (i.e., número de secuencias clasificadas) mediante el cálculo del centered log-ratio (CLR).
se_mb contiene una matriz llamada “refit”, la cual es una matriz de adyacencia dispersa (sparse adjacency matrix).
# Inferir la red usando SpiecEasi a partir de 'comau69_3'
# SpiecEasi cuenta con dos modelos de inferencia, que se definen con el argumento 'method': neighborhood selection ('mb') and inverse covariance selection ('glasso')
# Aquí, usamos method='mb' porque es mucho más rápido
# Úsa el argumento 'ncores' para indicar el número de procesadores disponible (si tienes más de uno)
se_mb <- spiec.easi(comau69_3, method='mb',
lambda.min.ratio=1e-2, nlambda=20,
pulsar.params=list(rep.num=50, ncores=3))
# Puedes exportar/guardar el objeto 'se_mb'
saveRDS(se_mb, "se_mb.RDS")- También, puedes descargar el objeto
se_mbAQUÍ y cargarlo a la sesión actual de R.
# Read network
se_mb <- readRDS(file = "se_mb.RDS")- Usamos las funciones
getRefit()yadj2igraph()del paquete SpiecEasi, para extraer la matriz refit del objetose_mby construir la red microbiana a partir de ella, respectivamente.
# Construir red a partir de la 'sparse adjacency matrix' o 'refit matrix'
# Add OTU names to rows and columns
# Create igraph objects
se_net <- adj2igraph(getRefit(se_mb),
rmEmptyNodes = TRUE, diag = FALSE,
vertex.attr = list(name = taxa_names(comau69_3))) # Usamos el ID de las taxas para nombrar los vértices o nodos de la red- El paquete phyloseq incluye la función
plot_network()para visualizar la red.
# Graficar la red 'se_net'
plot_network(se_net, comau69_3, type = "taxa", color = "Phylum", shape = "Kingdom", label = NULL)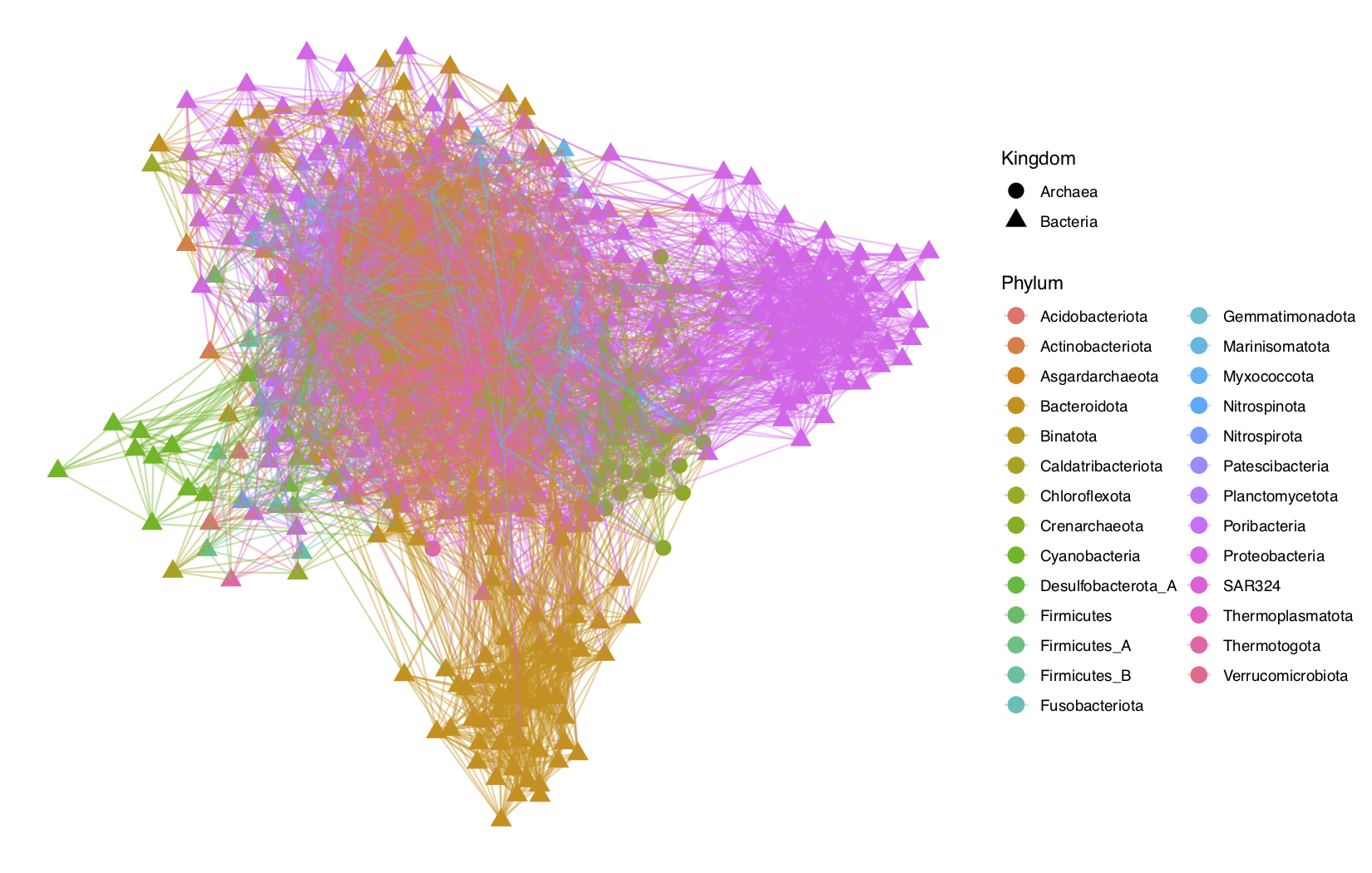
3.2 ggnet2
La función de phyloseq que usamos arriba para graficar la red, se basa en la función ggnet2 de ggplot2 para la visualización de redes en R. En este tutorial, vamos a usar ggnet2 porque consideramos más conveniente y fácil contar con la versatilidad y funciones de ggplot2 para visualizar y analizar la red.
- Para usar
ggnet2necesitamos un objeto de clase ‘network’ como input, el que obtenemos partir de la matriz de adyacencia de la red.
# Extraer matriz de adyacencia
net_class <- as_adjacency_matrix(se_net, type = "both")
# Generar objeto de clase 'network'
net_class <- network(as.matrix(net_class),
vertex.attrnames = taxa_names(comau69_3),
matrix.type = "adjacency", directed = F)- Visualizamos la red usando
ggnet2
ggnet2(net_class)
Seguramente notaste que no hay mucho que podamos deducir de esta visualización. Esto es porque la red representa las posibles relaciones entre todos los microorganismos detectados y clasificados en las muestras que componen el estudio. Inferir y graficar la red no es suficiente sino que solo el comienzo, lo que sigue es analizar las características y componentes de la red para estudiar hipótesis que busquen responder preguntas con respecto la comunidad microbiana.
4 Características de la red
En esta sección vamos a evaluar y visualizar las características de la red.
- Primero, vamos a crear una copia de la red, para poder volver atrás sin problema.
# Copiar el objeto 'se_net' a 'net'
net <- se_net- Características básicas de una red: vértices o nodos (nodes or vertex), bordes (edges), nombre de los nodos, y número total de nodos y bordes.
nodes <- V(net) # Nodos
edges <- E(net) # Bordes
node.names <- V(net)$name # Nombre de nodos
num.nodes <- vcount(net) # Número total de nodos
num.edges <- ecount(net) # Número total de bordes
# V() es por vértices o nodos
# E() es por edgesEn una red de co-ocurrencia microbiana; los nodos representan una taxa y los bordes o edges representan una relación entre las dos taxas que conecta.

4.1 Relaciones de co-ocurrencia positiva y negativa
La correlación entre dos taxas puede ser positiva o negativa:
Positiva: indica una relación directa o indirecta entre ambas taxas.
Negativa: indica una interacción competitiva o que las taxas no comparten nicho (non-overlapping niches).
- Cuántos edges positivos y negativos fueron inferidos por SpiecEasi.
# Extraer los coeficientes de regresión del objeto 'se_mb'
betaMat <- as.matrix(symBeta(getOptBeta(se_mb)))
# Calcular el número de edges positivos y negativos en la red
positive <- length(betaMat[betaMat>0])/2
negative <- length(betaMat[betaMat<0])/2
total <- length(betaMat[betaMat!=0])/2
# Dividimos por 2 ya que el edge esta representado por dos entradas en la matriz
# Puedes mirar los números en el panel 'Environment' de RStudio- Ahora, vamos a asignarle color a los edges positivos y negativos y a visualizar la red.
# El primer paso es extraer los signos de los coeficientes de regresión de la matriz de coeficientes de regresión
tax_ids <- taxa_names(comau69_3)
edges <- E(net) # edges
edge_colors <- c()
for(e_index in 1:length(edges)){
adj_nodes <- ends(net,edges[e_index])
xindex <- which(tax_ids==adj_nodes[1])
yindex <- which(tax_ids==adj_nodes[2])
beta <- betaMat[xindex,yindex]
if(beta>0){
edge_colors=append(edge_colors,"forestgreen") # positive
}else if(beta<0){
edge_colors=append(edge_colors,"red") # negative
}
}
E(net)$color <- edge_colors- Generamos el objeto de clase network y graficamos usando ggnet2.
# Extraer matriz de adyacencia
net_class <- as_adjacency_matrix(net, type = "both")
# Generar objeto de clase 'network'
net_class <- network(as.matrix(net_class),
vertex.attrnames = taxa_names(comau69_3),
matrix.type = "adjacency", directed = F)- Usamos el argumento
edge.colorde la funciónggnet2()para colorear los edges.
# Graficar red
ggnet2(net_class, edge.color = E(net)$color)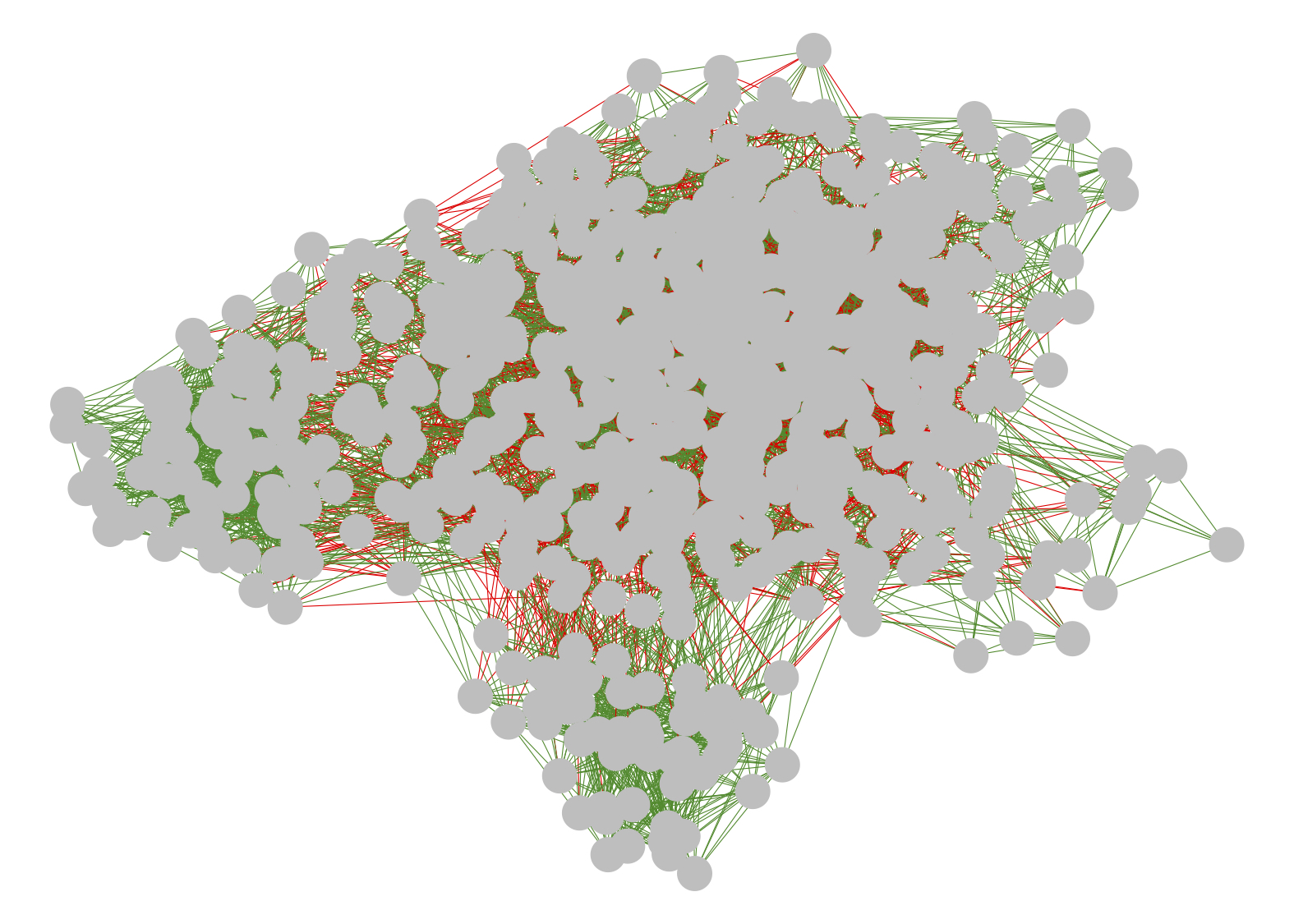
- Podemos hacer la visualización de la red más interesante agregando más información. Para ello, vamos a definir el nombre de cada nodo según su clasificación taxonómica a nivel de Phylum.
# Extraer tabla de taxonomía
tax_tbl <- as.data.frame(comau69_3@tax_table@.Data)
# Reemplazar el nombre de cada nodo por su Phylum
V(net)$name <- as.character(getTaxonomy(V(net)$name, tax_tbl, level = "phylum", useRownames = TRUE))
# Guardar la lista de Phylum por nodo en 'nodenames'
nodenames <- V(net)$name
# Extraer matriz de adyacencia
net_class <- as_adjacency_matrix(net, type = "both")
# Generar objeto de clase 'network'
net_class <- network(as.matrix(net_class),
vertex.attrnames = taxa_names(comau69_3),
matrix.type = "adjacency", directed = F)# Graficar red
ggnet2(net_class, color = nodenames, edge.color = E(net)$color)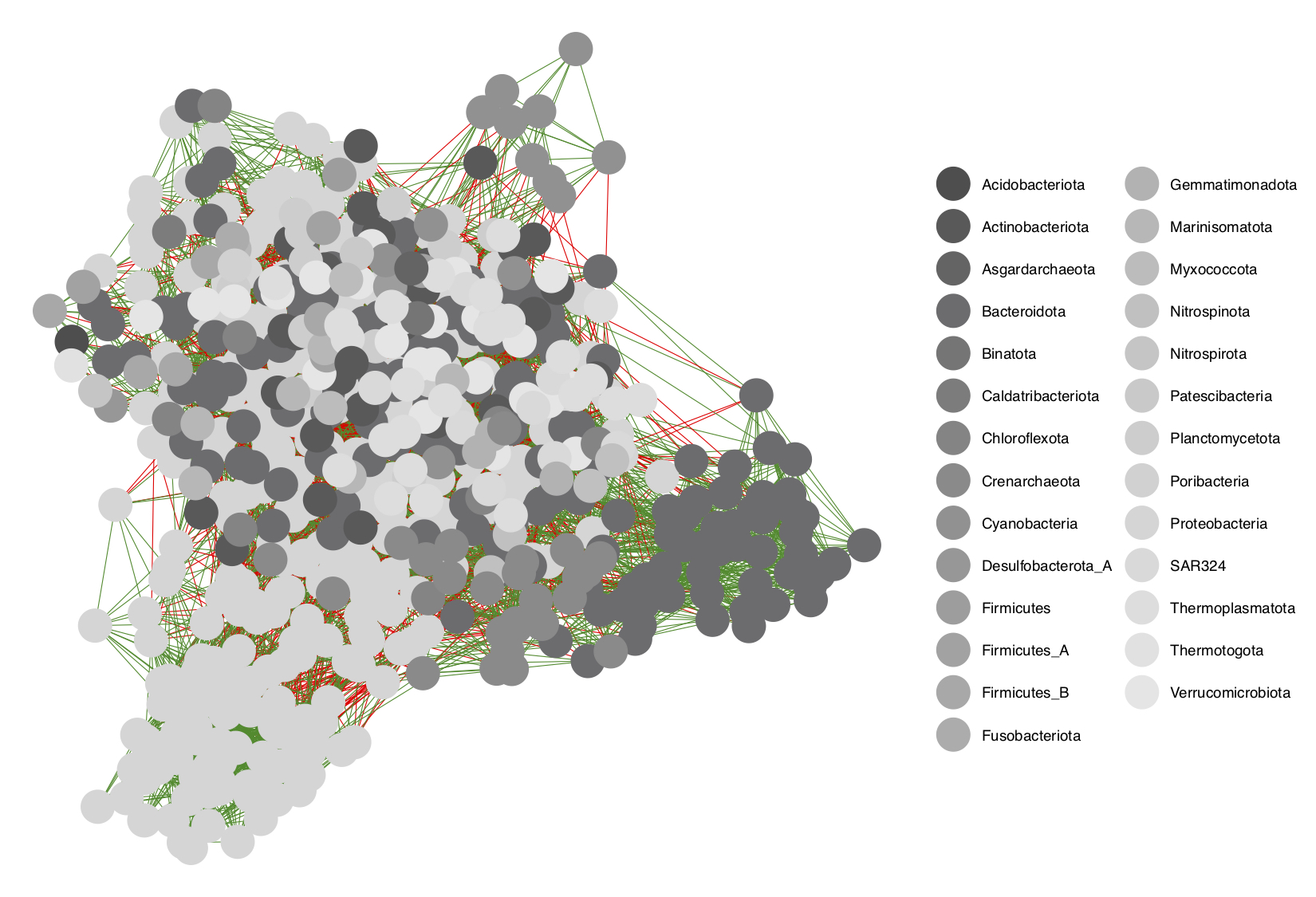
- Finalmente, personalizamos la visualización asignando colores personalizados a los nodos según su Phylum.
# Definir paleta de colores por Phylum
colors1 <- c("Acidobacteriota" = "#263238",
"Actinobacteriota" = "#bf360c",
"Asgardarchaeota" = "#3690c0",
"Bacteroidota" = "#8c96c6",
"Binatota" = "#E91E63",
"Caldatribacteriota" = "#2196F3",
"Chloroflexota" = "#009688",
"Crenarchaeota" = "#FF9800",
"Cyanobacteria" = "#795548",
"Desulfobacterota_A" = "#91003f",
"Dadabacteria" = "#4a148c",
"Firmicutes" = "#673AB7",
"Firmicutes_A" = "#03A9F4",
"Firmicutes_B" = "#33691e",
"Fusobacteriota" = "#FF5722",
"Gemmatimonadota" = "#3F51B5",
"k__Bacteria" = "#CDDC39",
"Latescibacterota" = "#673AB7",
"k__Bacteria" = "#607D8B",
"Marinisomatota" = "#00BCD4",
"Myxococcota" = "#FFEB3B",
"Nanoarchaeota" = "#607D8B",
"Nitrospinota" = "#F44336",
"Nitrospirota" = "#c7e9b4",
"Patescibacteria" = "#4CAF50",
"Planctomycetota" = "#9C27B0",
"Poribacteria" = "#8c2d04",
"Proteobacteria" = "#FFC107",
"SAR324" = "#9E9E9E",
"Thermoplasmatota" = "#ffd180",
"Thermotogota" = "#e7298a",
"Verrucomicrobiota" = "#37474f")ggnet2(net_class, color = nodenames, palette = colors1, edge.color = E(net)$color)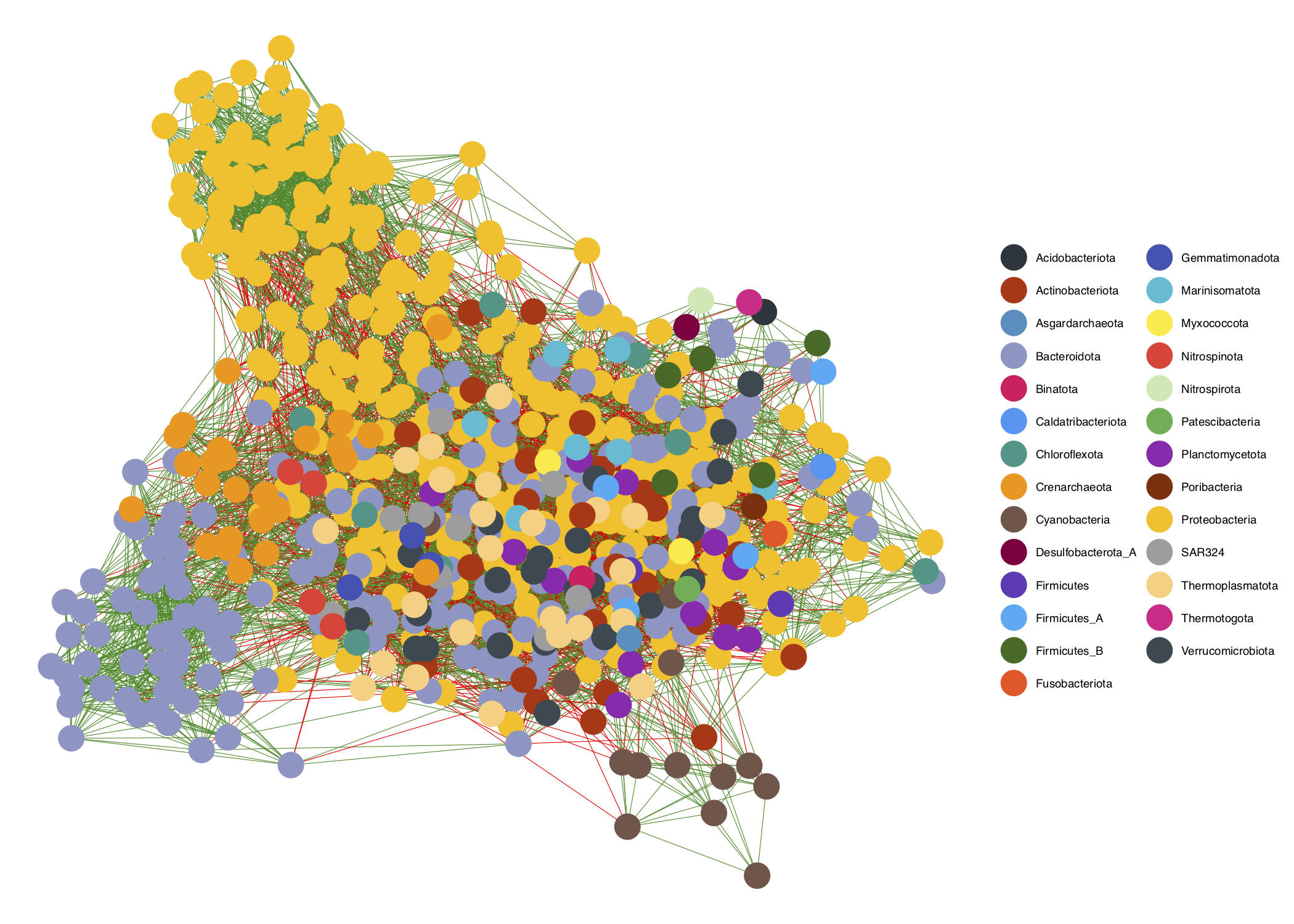
4.2 Estabilidad visual
Cada vez que graficamos una red, las posiciones de los nodos son re-calculadas. Para tener una representación visualmente estable y comparable de la red, podemos usar coordinadas fijas para todos los nodos y asignar el diseño como un atributo fijo a la red.
- Fijar el diseño de la red.
# Las coordenadas deben estar en forma de una matriz n vs. m, donde n es el nombre de los nodos y m es las coordenadas x e y de cada nodo
# Asignar coordenadas al diseño de la red
net$layout <- array(1:40, dim = c(20, 2))
# Asignar el diseño como un atributo fijo de la red
net$layout <- layout.fruchterman.reingold(net)Después de fijar el diseño de la red, solo hace falta agregar el argumento mode = net$layout a la función ggnet2() cada vez que queramos graficar la red y la distribución de los nodos se mantendrá igual.
# Graficar red
ggnet2(net_class, mode = net$layout)Usaremos esta estrategia en el resto del tutorial.
5 Estadística de la red
En esta sección vamos a calcular node degree, node centrality, y transitivity de la red.
- Primero, creamos una copia de la red y fijamos el diseño.
# Copiar el objeto 'se_net' a 'net'
net <- se_net
# Asignar coordenadas al diseño de la red
net$layout <- array(1:40, dim = c(20, 2))
# Asignar el diseño como un atributo fijo de la red
net$layout <- layout.fruchterman.reingold(net)5.1 Degree
El valor de degree de un nodo representa el número de edges conectados al nodo en cuestión, es decir, mientras más relaciones tenga un nodo en particular con otros nodos en la red mayor degree.
La distribución de valores de degree de los nodos que componen una red nos da una idea de la conectividad de los nodos en la red.
- Usamos el paquete igraph para calcular los valores de degree y su distribución en la red.
# Calcular node degree
deg <- igraph::degree(net, mode = "all")
# Calcular degree distribution
deg.dist <- degree_distribution(net, mode = "all", cumulative = F)- Graficar distribución de los valores de degree de los nodos en la red.
# Graficar degree distribution
plot(deg.dist, xlab = "Nodes degree", ylab = "Probability")
lines(deg.dist)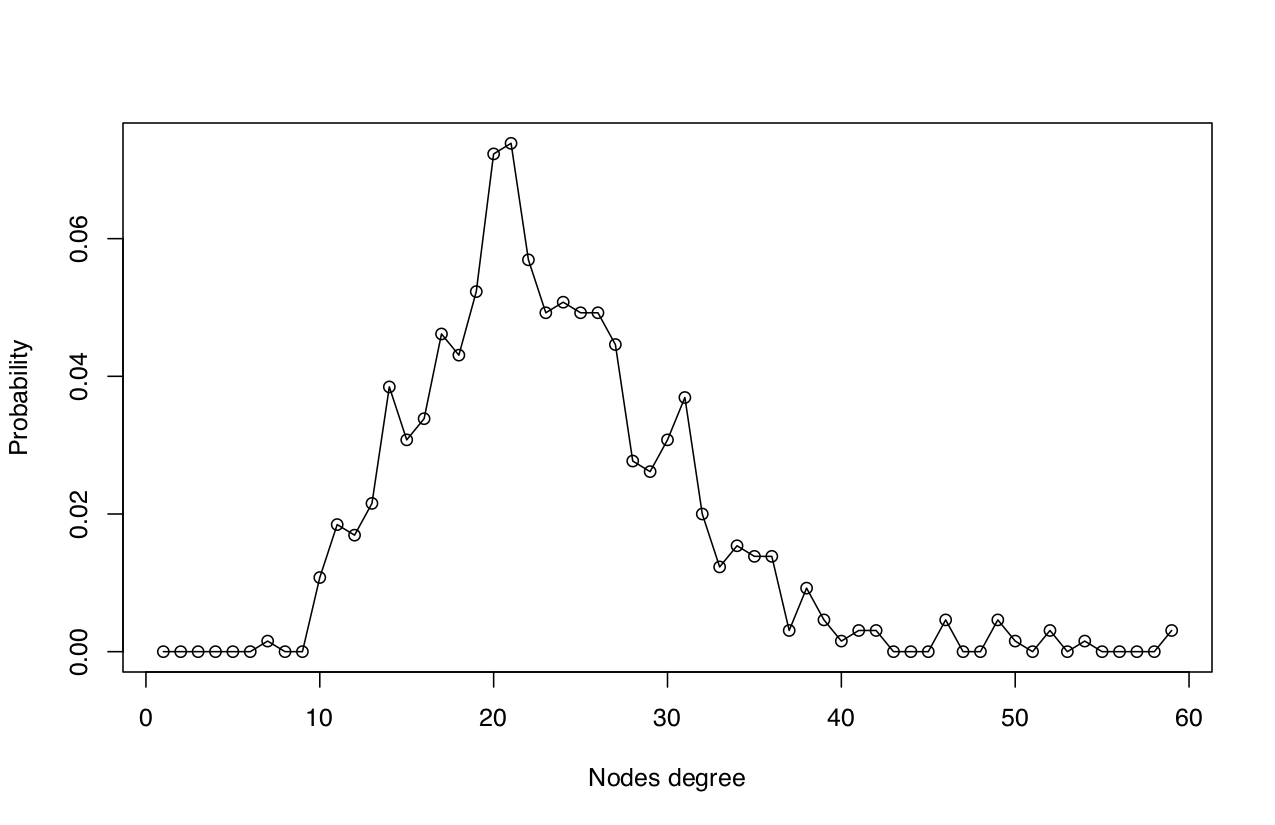
5.2 Node centrality
La centralidad de los nodos en la red se mide de dos formas: closeness y betweenness.
5.2.1 Closeness
Closeness se refiere a que tan central es cada nodo para la red completa. Para medir closeness se recorre la red de forma aleatoria y se cuantifica la frecuencia con la que se visita cada nodo. Nodos que son visitados con mayor frecuencia tienen mayor valor de closeness.
- Usamos la función
closeness()del paquete igraph para calcular los valores de closeness de todos los nodos.
clos <- igraph::closeness(net, mode = "all")5.2.2 Betweenness
Betweenness también es una medida de centralidad de los nodos que componen la red. El valor de betweenness de un nodo se calcula como el número total de rutas más cortas desde todos los nodos a todos los otros nodos que pasan a través del nodo en cuestión. Nodos que presentan un valor de betweenness alto, son aquellos que conectan grupos de nodos que soportan la red.
Un nodo puede tener un degree bajo y un betweenness alto, esto representaría un nodo que tiene pocas conexiones, pero estas conexiones conectan nodos altamente conectados con otros nodos.
- Usamos la función
betweenness()del paquete igraph para calcular los valores de betweenness de todos los nodos en la red.
betw <- igraph::betweenness(net, v = V(net))
# Si quieres calcular betweenness para uno o varios nodos en particular, usa el argumento 'v' para definir los nodos de interés- Podemos usar la función
centralityPlot()del paquete qgraph para graficar los valores de betweenness, closeness y degree (eje x) de todos los nodos (eje y) en la red.
centralityPlot(net, include = c("Betweenness", "Closeness", "Degree")) +
theme(axis.text.y = element_blank())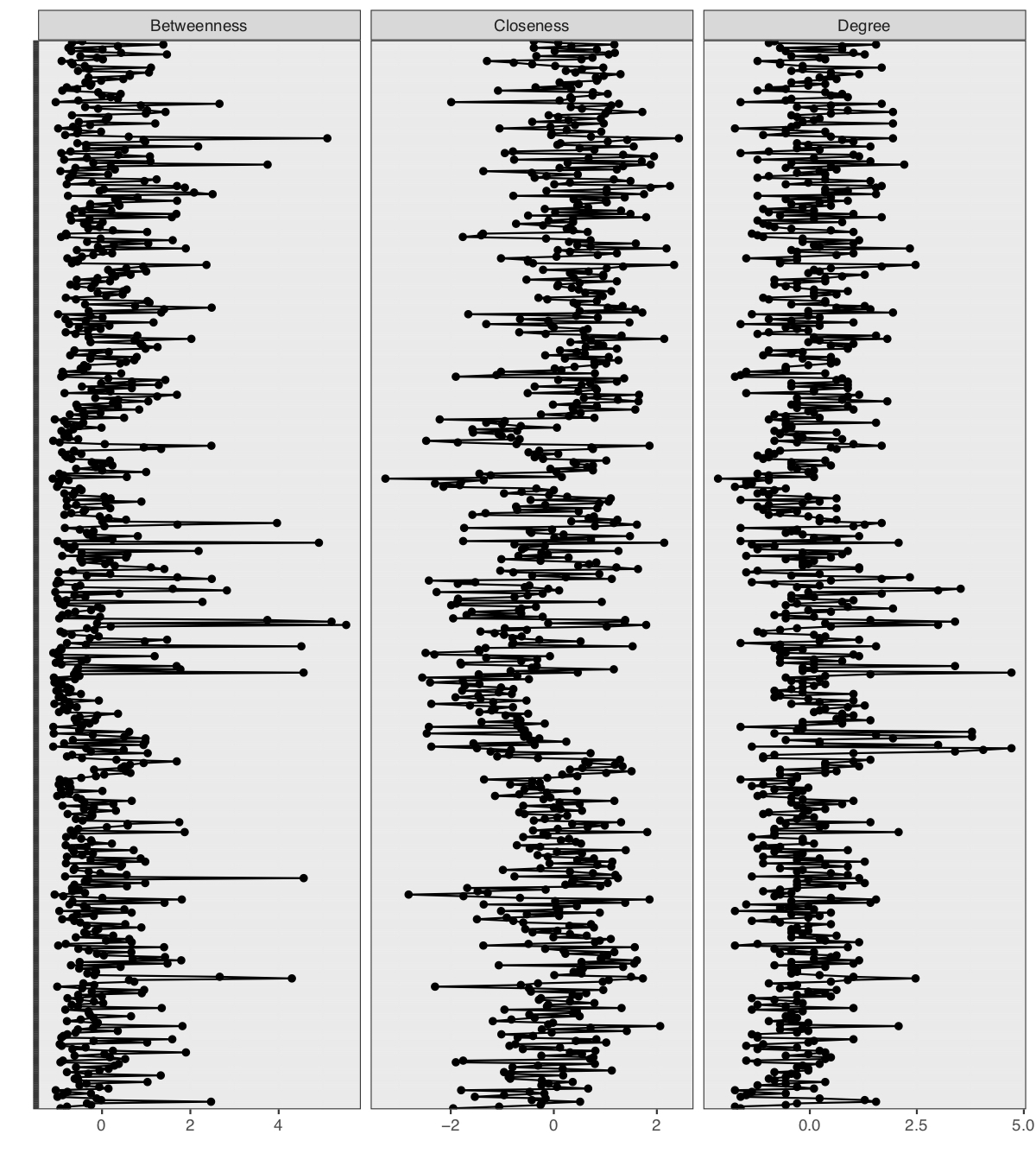
5.3 Transitivity
Transitivity, también llamado coeficiente de agrupamiento (clustering coefficient), mide la probabilidad de que nodos adyacentes al nodo en cuestión esten conectados entre sí.
- Usamos la función
transitivity()del paquete igraph para calcular transitivity.
# Usamos el argumento type = "global" para calcular el valor de transitivity total para toda la red
clustering_coeff_global <- transitivity(net, type = "global")
# Usamos el argumento type = "local" para calcular el valor de transitivity de cada nodo en la red
clustering_coeff_local <- transitivity(net, type = "local")5.4 Average Nearest Neighbor Degree (ANND)
ANND nos permite evaluar si la correlación entre los valores de degree de nodos vecinos es positiva o negativa. Si es positiva; los nodos con alto degree tienden a conectarse con otros nodos con alto degree. Mientras que, si es negativa; los nodos con alto degree tienden a conectarse con nodos con bajo degree.
Calcular ANND puede ser útil al momento de querer analizar cierto(s) nodo(s) de interés y su relación con nodos adyacentes.
- Usamos la función
knn()del paquete igraph para calcular ANND.
# Para calcular el valor de ANND para todos los nodos en la red, usamos el argumento vids = V(net)
net.knn <- knn(net, vids = V(net))
head(net.knn$knn)## TI_27683 TI_39035 TI_27681 TI_13593 TI_13597 TI_20829
## 24.53333 24.22857 26.15385 22.76000 23.80769 25.32258# Si quieres calcular ANND para uno o varios nodos en particular, usa el argumento 'vids' para definir los nodos de interés6 Estructura de la red
En esta sección vamos a evaluar la estructura de la red, nos referimos a la detección de módulos o clusters dentro de la red y a la extracción de estos para analizarlos como una red independiente.
- Primero, creamos una copia de la red y fijamos el diseño.
# Copiar el objeto 'se_net' a 'net'
net <- se_net
# Asignar coordenadas al diseño de la red
net$layout <- array(1:40, dim = c(20, 2))
# Asignar el diseño como un atributo fijo de la red
net$layout <- layout.fruchterman.reingold(net)6.1 Detección de módulos
En una red de co-ocurrencia microbiana pueden existir grupos de nodos que conformen un módulo o cluster, como una sub-red dentro de la red. Un módulo o cluster se define como un conjunto de nodos que están fuertemente relacionados entre sí, y a su vez, todos ellos se relacionan en menor medida con nodos que no pertenecen al grupo.
It should be noted that igraph methods output an object containing various information on the detected clusters, including but not limited to the membership of each cluster (membership function below)
- Usamos las funciones
walktrap.community()ymembership()del paquete igraph para, primero detectar posibles módulos dentro de la red, y segundo para consultar la membresía de cada nodo (qué nodo pertenece a qué módulo).
# Esta función intenta detectaar sub-redes densamente conectadas, usando 'random walks'
# 'random walks' se refiere a "recorrer" la red de forma aleatoria
# Se supone que "recorridos cortos" tienden a quedarse en la misma sub-red o módulo
wt <- walktrap.community(net)
# Consultar membresía de cada nodo
membership(wt) %>% head()## TI_27683 TI_39035 TI_27681 TI_13593 TI_13597 TI_20829
## 6 1 2 3 3 3- Usando la función
plot_dendrogram()podemos visualizar los módulos o clusters detectados en un dendograma.
# Visualizar la estructura jerárquica de la comunidad microbiana en un dendograma
igraph::plot_dendrogram(wt)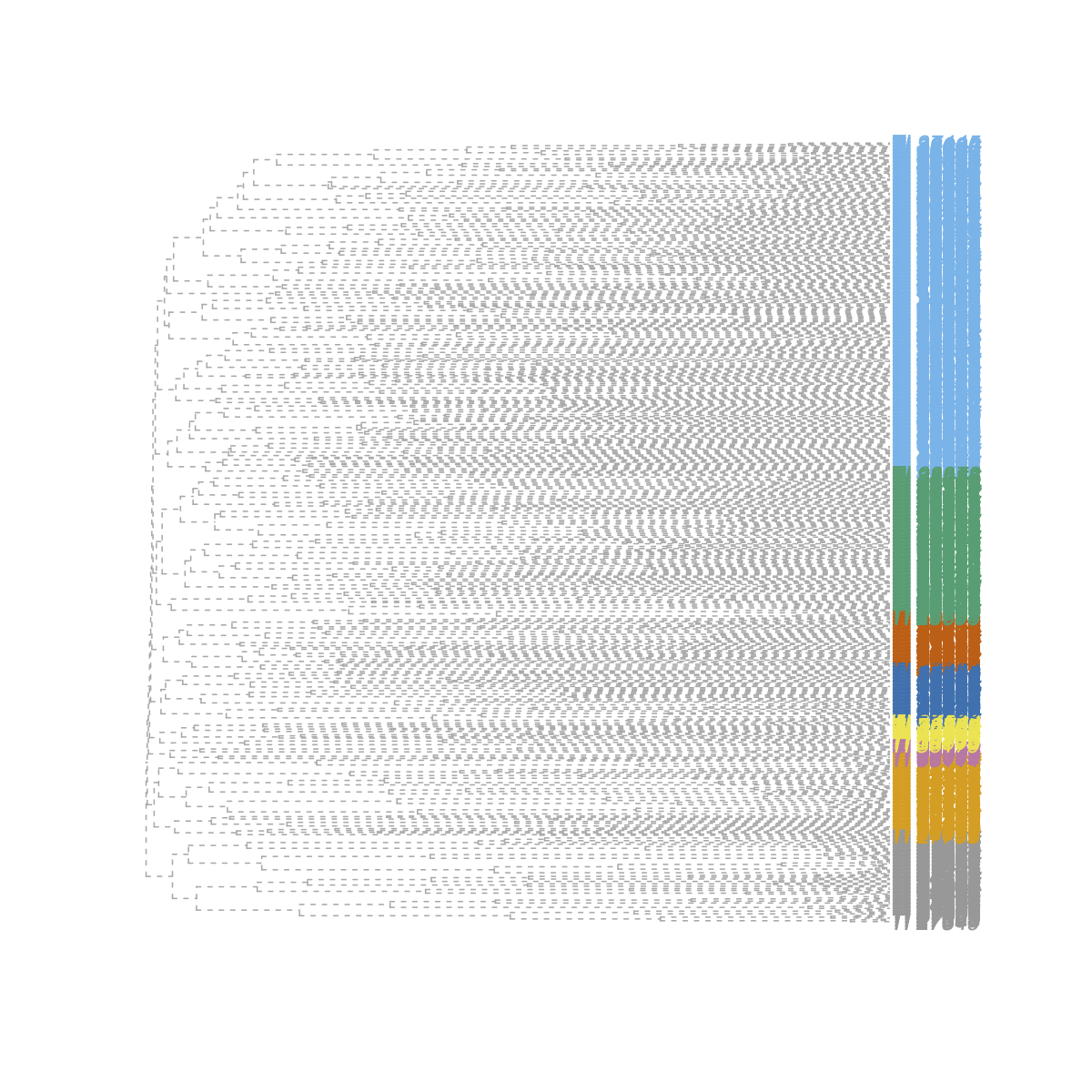
6.1.1 Modularidad
Modularidad es una buena medida de la fuerza de división de una red en módulos o clusters. Una alta modularidad indica que la red presenta densas conexiones dentro de ciertos grupos de nodos (módulos), y a su vez, conexiones dispersas entre grupos de nodos diferentes.
- La modularidad de una red calculada con respecto a la membresía de los nodos que la componen, puede ser usada para estimar qué tan separados estan los módulos uno de otro.
# Calcular modularidad
modularity(net, membership(wt))## [1] 0.41691916.1.2 Visualizar módulos
- Visualizar módulos o clusters en la red
# Plot
plot(wt, net)
Nuevamente, vamos a utilizar ggnet2 para visualizar la red y sus estructura agregando más información para un análisis más exhaustivo de la comunidad microbiana.
- Generar objeto de clase network y visualizar la red definiendo diseño y coloreando los nodos según el módulo al que pertenecen.
# Extraer matriz de adyacencia
net_class <- as_adjacency_matrix(net, type = "both")
# Generar objeto de clase 'network'
net_class <- network(as.matrix(net_class),
vertex.attrnames = taxa_names(comau69_3),
matrix.type = "adjacency", directed = F)ggnet2(net_class, mode = net$layout, color = wt$membership)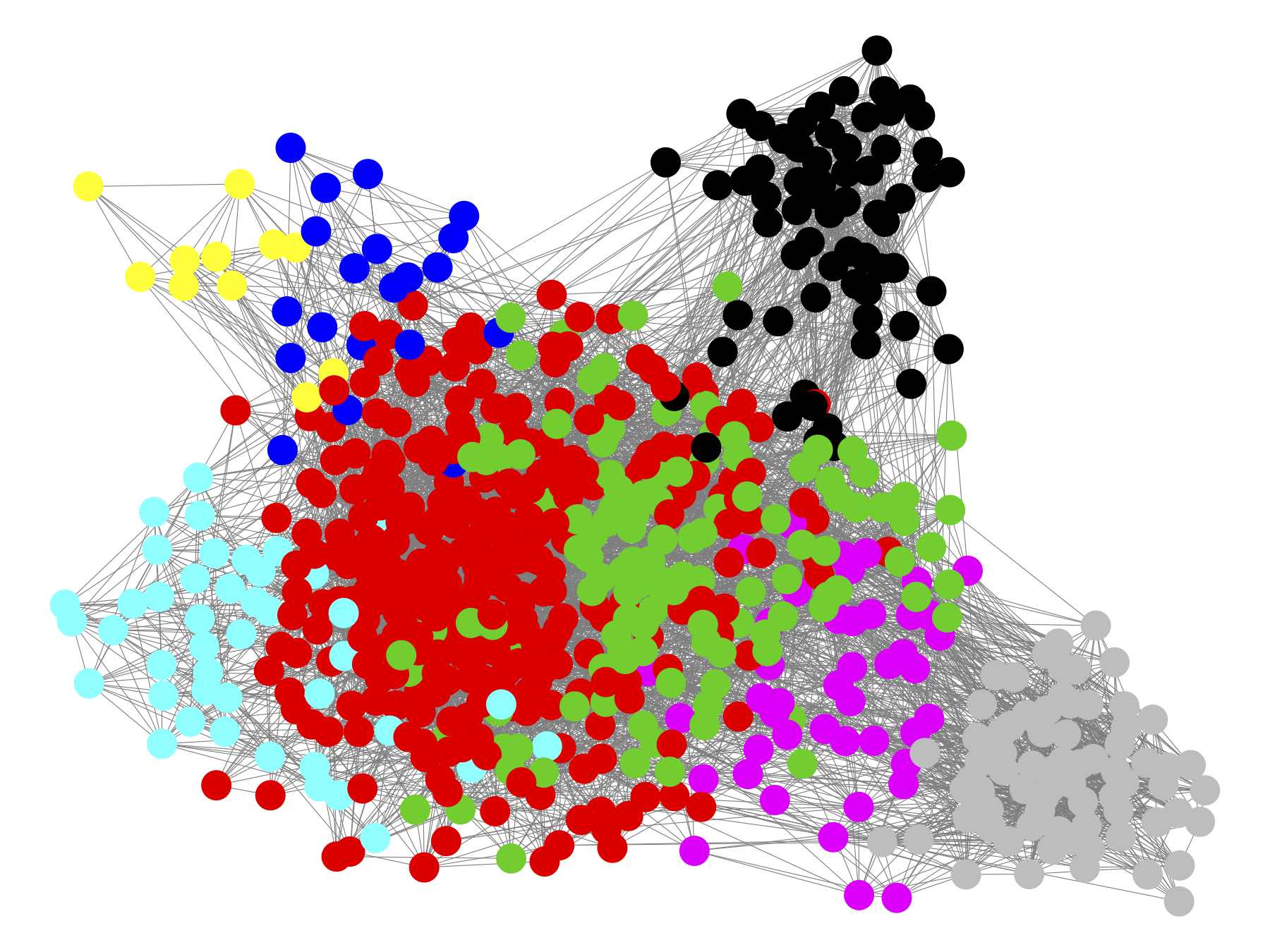
- Ahora, vamos a usar la función
stat_ellipse()de ggplot2 para dibujar una elipse, por módulo, alrededor del grupo de nodos miembros de cada módulo en particular.
ggnet2(net_class, mode = net$layout, color = wt$membership) +
stat_ellipse(aes(color = factor(wt$membership)), type = "norm")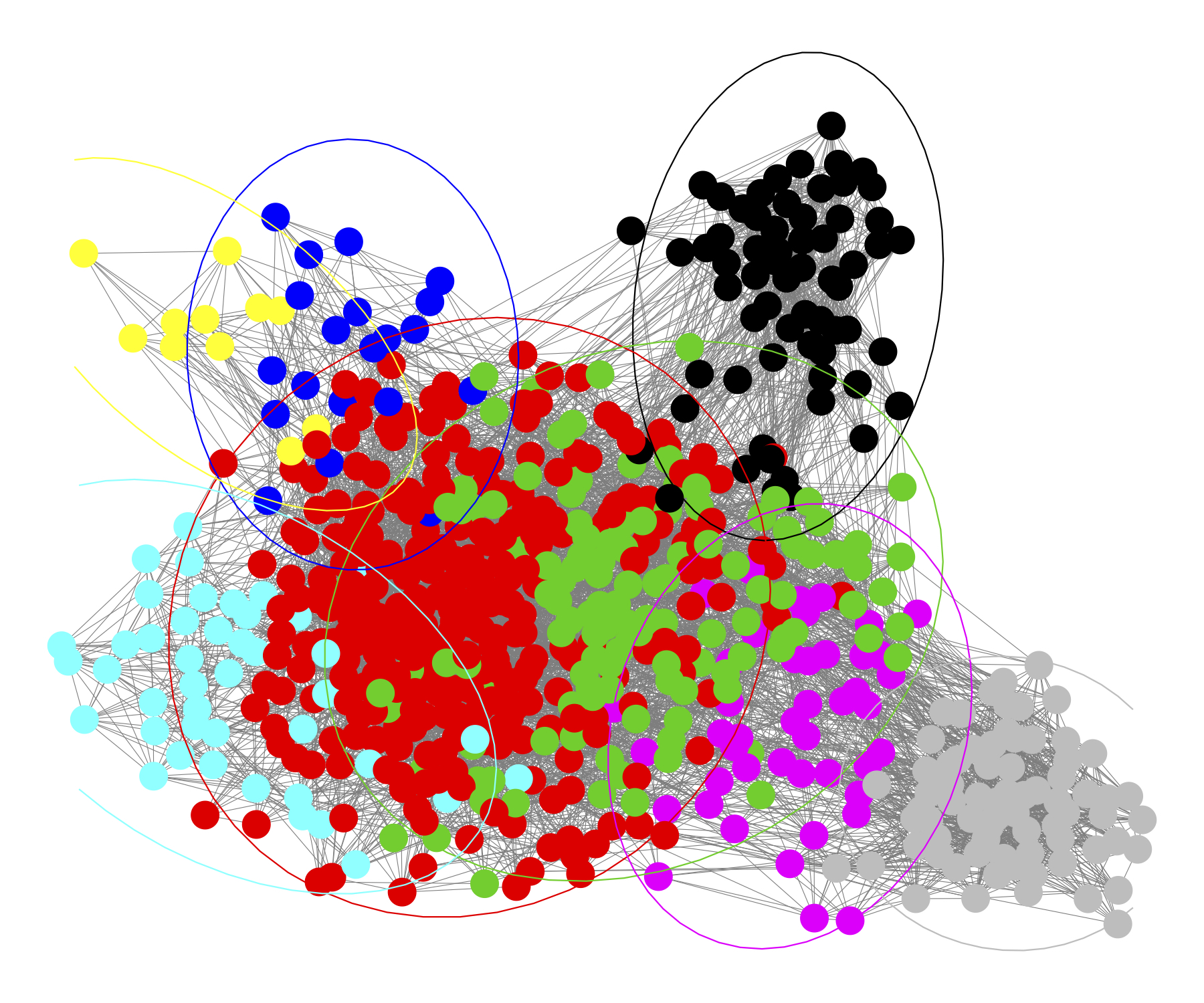
- Finalmente, vamos a enriquecer la visualización de la red, usando todas las habílidades de visualización de ggnet2 que hemos practicado hasta ahora.
# Extraer la clasificación taxonómica de cada nodo a nivel de Phylum
nodenames <- as.character(getTaxonomy(V(net)$name, tax_tbl, level = "phylum", useRownames = TRUE))- Graficamos la red de co-ocurrencia microbiana; coloreando los nodos según su taxonomía a nivel de Phylum (usando una paleta de colores personalizada), coloreando los edges según si representan una relación positiva o negativa, y definiendo los módulos o clusters dentro de la red usando elipses.
ggnet2(net_class, mode = net$layout, color = nodenames, palette = colors1, edge.color = edge_colors) +
stat_ellipse(aes(group = factor(wt$membership)), type = "norm")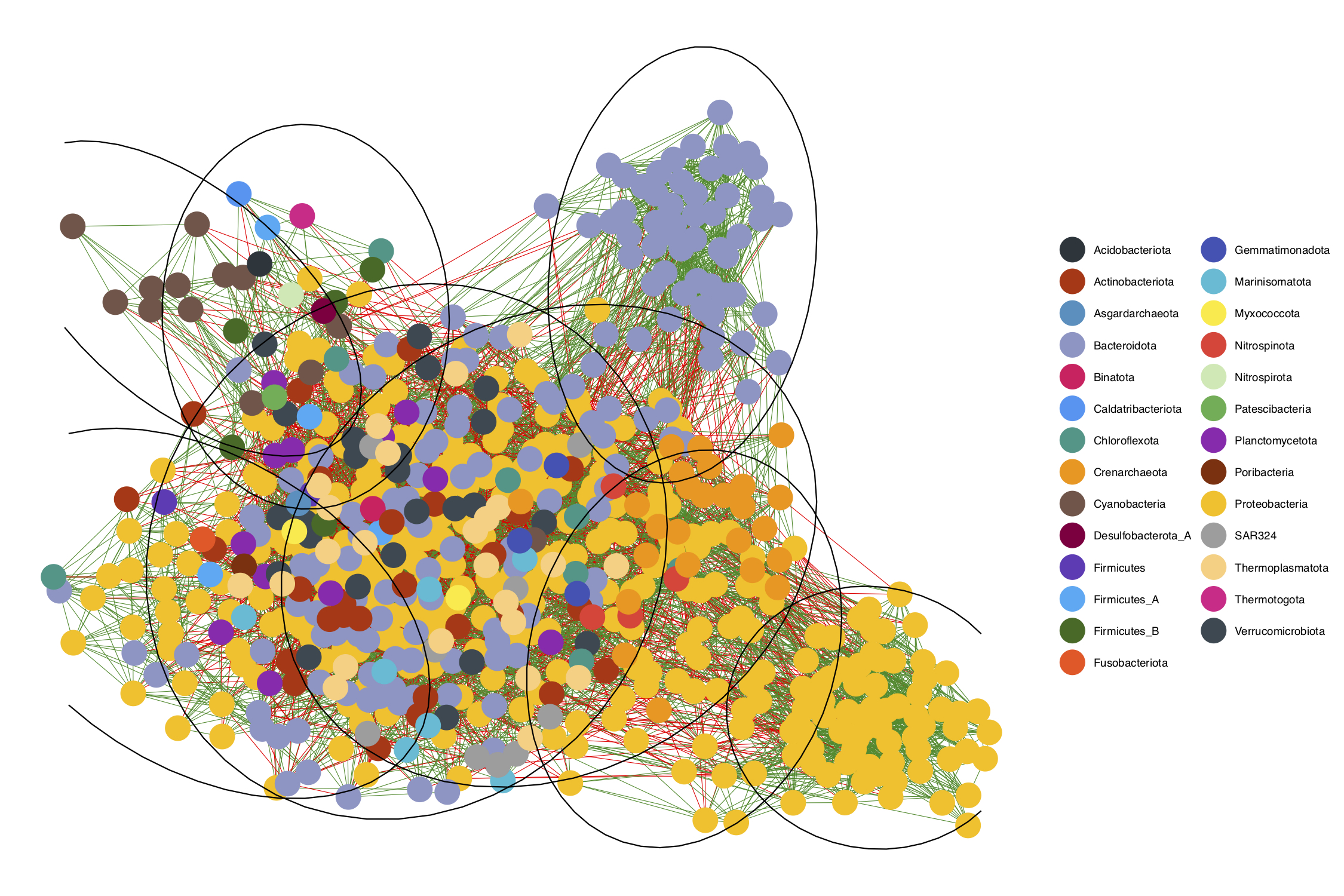
6.2 Sub-redes
El paquete igraph incluye la función induced_subgraph() que nos permite extraer o inducir sub-redes desde la red original. Por supuesto, podemos usar esta función para extraer los módulos que detectamos arriba y analizarlos uno a la vez. De hecho, cualquier selección de nodos puede ser utilizada para extraer o inducir una sub-red.
Cabe señalar que todos los análisis aplicables a una red pueden ser aplicados a una sub-red.
- Primero, creamos un objeto con los nodos pertenecientes al módulo 1 y/o 2, y luego, usamos este objeto para inducir la sub-red.
# Extraer nodos miembros del módulo 1
m1 <- V(net)[wt$membership == 1]
# Inducir sub-red 'm1': extraer módulo 1
m1_subnet <- induced_subgraph(net, m1)
# Extraer nodos miembros del módulo 2
m2 <- V(net)[wt$membership == 2]
# Inducir sub-red 'm1': extraer módulo 2
m2_subnet <- induced_subgraph(net, m2)- Ahora que hemos extraído el módulo 1 “m1” de la red, y lo tenemos en el objeto
m2_subnetcomo una red aparte, podemos visualizarlo de forma independiente.
# Comenzamos fijando el diseño de la red m1
# Asignar coordenadas al diseño de la red
m1_subnet$layout <- array(1:40, dim = c(20, 2))
# Asignar el diseño como un atributo fijo de la red
m1_subnet$layout <- layout.fruchterman.reingold(m1_subnet)
# Creamos el objeto de clase network (input necesario para ggnet2)
# Extraer matriz de adyacencia
m1_class <- as_adjacency_matrix(m1_subnet, type = "both")
# Generar objeto de clase 'network'
m1_class <- network(as.matrix(m1_class),
matrix.type = "adjacency", directed = F)- Graficamos la sub-red correspondiente al módulo 1.
ggnet2(m1_class, mode = m1_subnet$layout)
Tal y como lo hemos hecho antes; podemos enriquecer la visualización de la sub-red agregando más información acerca de los nodos y edges del módulo 1.
- Extraemos la clasificación taxonómica a nivel de familia de los nodos miembros del módulo 1.
m1_nodenames <- as.character(getTaxonomy(V(m1_subnet)$name, tax_tbl, level = "family", useRownames = TRUE))- Graficamos el módulo 1, coloreando los nodos según a qué familia pertenecen.
ggnet2(m1_class, mode = m1_subnet$layout, color = m1_nodenames)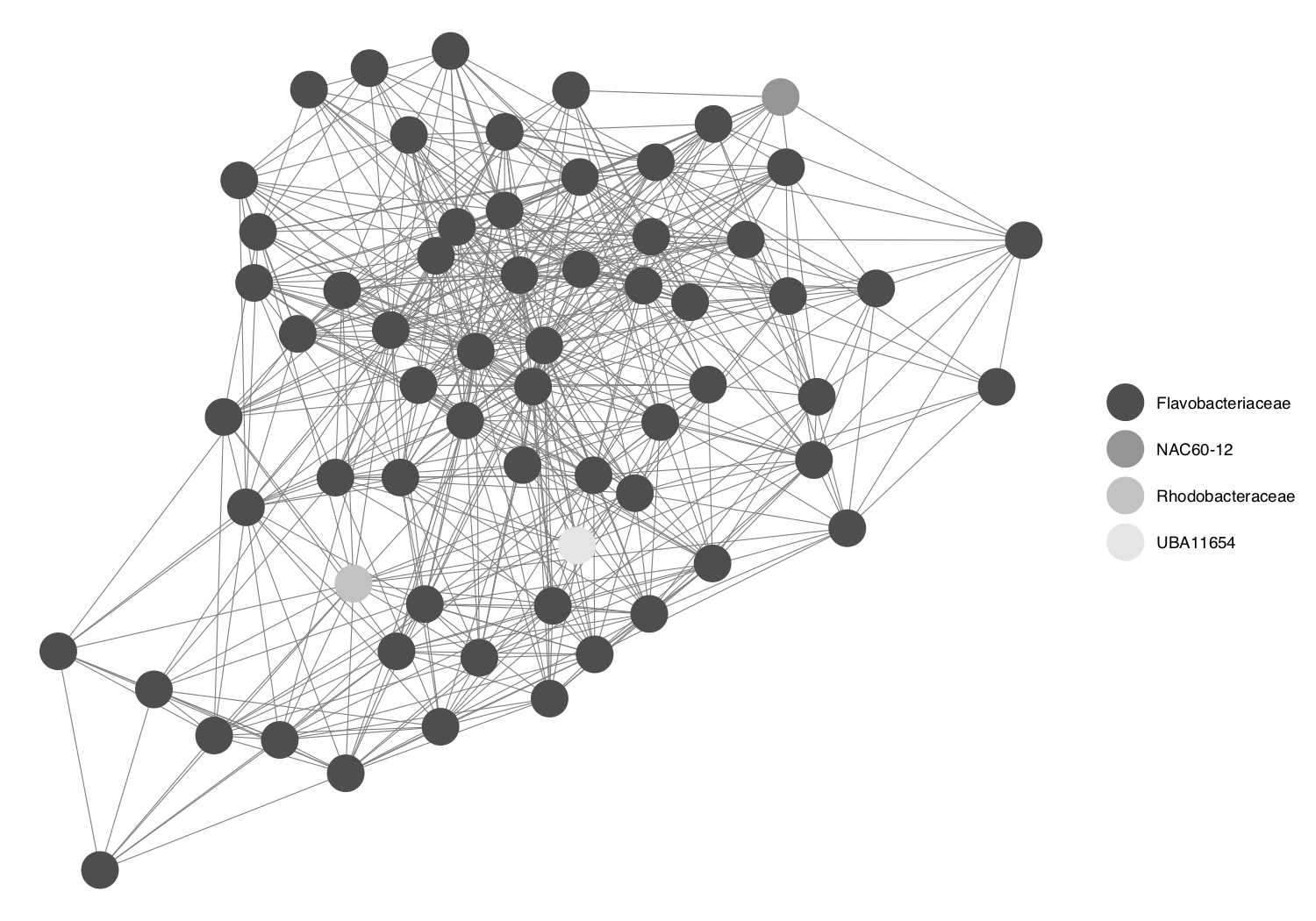
- Identificamos las “familias” presentes en el módulo 1 y creamos una paleta de colores personalizada.
unique(m1_nodenames)## [1] "Rhodobacteraceae" "UBA11654" "Flavobacteriaceae"
## [4] "NAC60-12"colors2 <- c("Rhodobacteraceae" = "#BF0B3B",
"UBA11654" = "#1835D9",
"Flavobacteriaceae" = "#F2B90C",
"NAC60-12" = "#238C2A")- Finalmente, graficamos el módulo 1 coloreando los nodos según la familia a la que pertenecen.
ggnet2(m1_class, mode = m1_subnet$layout, color = m1_nodenames, palette = colors2)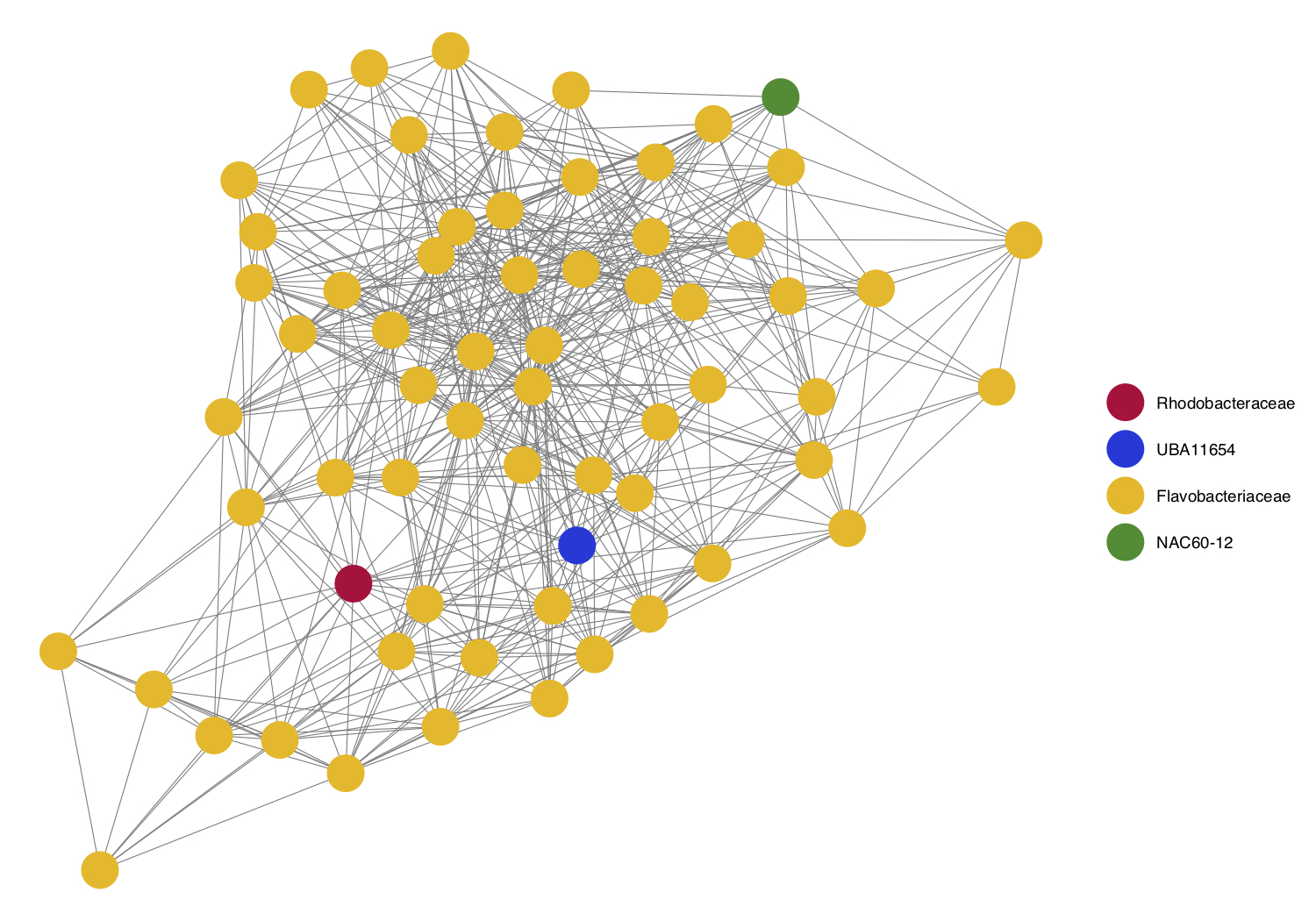
También podemos evaluar otras características de la sub-red, así:
vcount(m1_subnet) # vertices## [1] 64ecount(m1_subnet) # edges## [1] 581vcount(m2_subnet) # vertices## [1] 276ecount(m2_subnet) # edges## [1] 2279transitivity(m1_subnet)## [1] 0.4706745transitivity(m2_subnet)## [1] 0.14614157 Búsqueda de keystone species
 Una de las aplicaciones más importantes de inferir la red de co-ocurrencia microbiana es la búsqueda de; hub species y keystone species. Nodos identificados como especies hub o keystone representan taxas potencialmente centrales y con un rol importante dentro de la comunidad microbiana en estudio.
Una de las aplicaciones más importantes de inferir la red de co-ocurrencia microbiana es la búsqueda de; hub species y keystone species. Nodos identificados como especies hub o keystone representan taxas potencialmente centrales y con un rol importante dentro de la comunidad microbiana en estudio.
Aquellos nodos con los valores de degree más altos se identifican como hub species, que representan taxas altamente conectadas dentro de la comunidad microbiana.
Una hub specie podría representar una keystone specie. Keystone species se consideran muy importantes para la estructura y funcionamiento de la comunidad microbiana, y su remoción podría causar el colapso de la comunidad.
¿Cómo podemos evaluar si una hub specie es una keystone specie?
 Node Centrality
Node Centrality
Una hub specie es detectada por su alto degree en comparación a todas las otras taxas/nodos en la comunidad microbiana/red. Luego, al estimar node centrality calculando betweenness establecemos además que la taxa/nodo es central dentro de la comunidad microbiana/red, ya que conecta grupos de taxas/nodos que soportan a la comunidad/red.
Como mencionamos antes (sección 5); un nodo puede tener un degree bajo y un betweenness alto, esto representaría un nodo que tiene pocas conexiones, pero estas conexiones conectan nodos altamente conectados con otros nodos. Por lo tanto, podrían representar una keystone specie, ya que removerlos podría causar el colapso de la comunidad.
También, podemos acumular evidencia para definir un nodo como representante de una keystone specie dentro de la comunidad microbiana, haciendo el experimento de remover el nodo de la red y ver qué pasa (i.e., qué tanto afecta a la estructura de la red).
 Sin embargo, en este tutorial vamos a realizar la búsqueda de keystone species mediante el cálculo de degree y betweenness, seleccionando aquellos nodos que presenten los valores más altos en comparación a todos los nodos en la red.
Sin embargo, en este tutorial vamos a realizar la búsqueda de keystone species mediante el cálculo de degree y betweenness, seleccionando aquellos nodos que presenten los valores más altos en comparación a todos los nodos en la red.
7.1 Node degree
- Calculamos los valores de degree de todos los nodos en la red y ordenamos de mayor a menor.
# Calcular degree
deg <- igraph::degree(net)
# Ordenar nodos según degree de mayor a menor
deg_sort <- sort(deg, decreasing = TRUE)
head(deg_sort) # Mira las primeras 6 filas del objeto 'deg_sort'## TI_27397 TI_27345 TI_27344 TI_27357 TI_27360 TI_27772
## 58 58 53 51 51 49- Graficamos un histograma para visualizar los valores de degree de los nodos que componen la red.
# Transformar 'deg_sort' a un objeto de tipo 'data frame', necesario para graficar el histograma
deg_df <- as.data.frame(deg_sort)# Graficar histograma
# También usamos la función `geom_vline()` del paquete ggplot2 para dibujar una línea discontinua para marcar la media o promedio de los valores de degree
ggplot(deg_df, aes(x = deg_sort)) +
geom_histogram(binwidth = 1) +
scale_x_continuous(breaks=c(0,10,20,30,40,50,60)) +
geom_vline(aes(xintercept=mean(deg_sort)),
color="black", linetype="dashed", size=1) +
theme_minimal() +
labs(x = "Degree", y = "Node count")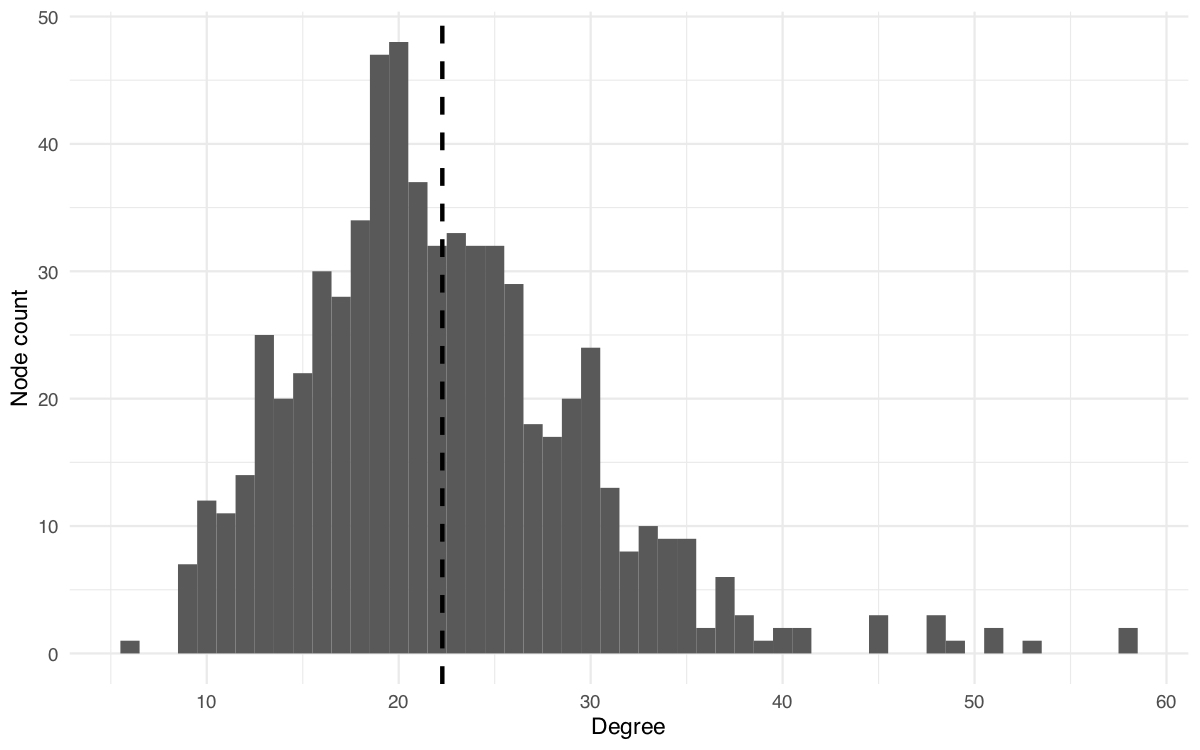
7.2 Node centrality
- Como medida de centralidad, calculamos los valores de betweenness de todos los nodos en la red y ordenamos de mayor a menor.
# Calcular betweenness
bn <- igraph::betweenness(net)
# Ordenar nodos según betweenness de mayor a menor
bn_sort <- sort(bn, decreasing = TRUE)
head(bn_sort) # Mira las primeras 6 filas del objeto 'bn_sort'## TI_27683 TI_27698 TI_38980 TI_29620 TI_23094 TI_27397
## 3184.922 3026.017 2980.506 2889.191 2724.146 2723.078- Graficamos un histograma para visualizar los valores de betweenness de los nodos que componen la red.
# Transformar 'bn_sort' a un objeto de tipo 'data frame', necesario para graficar el histograma
bn_df <- as.data.frame(bn_sort)# Graficar histograma
# También usamos la función `geom_vline()` del paquete ggplot2 para dibujar una línea discontinua para marcar la media o promedio de los valores de betweenness
ggplot(bn_df, aes(x = bn_sort)) +
geom_histogram(binwidth = 30) +
geom_vline(aes(xintercept=mean(bn_sort)),
color="black", linetype="dashed", size=1) +
theme_minimal() +
labs(x = "Betweenness", y = "Node count")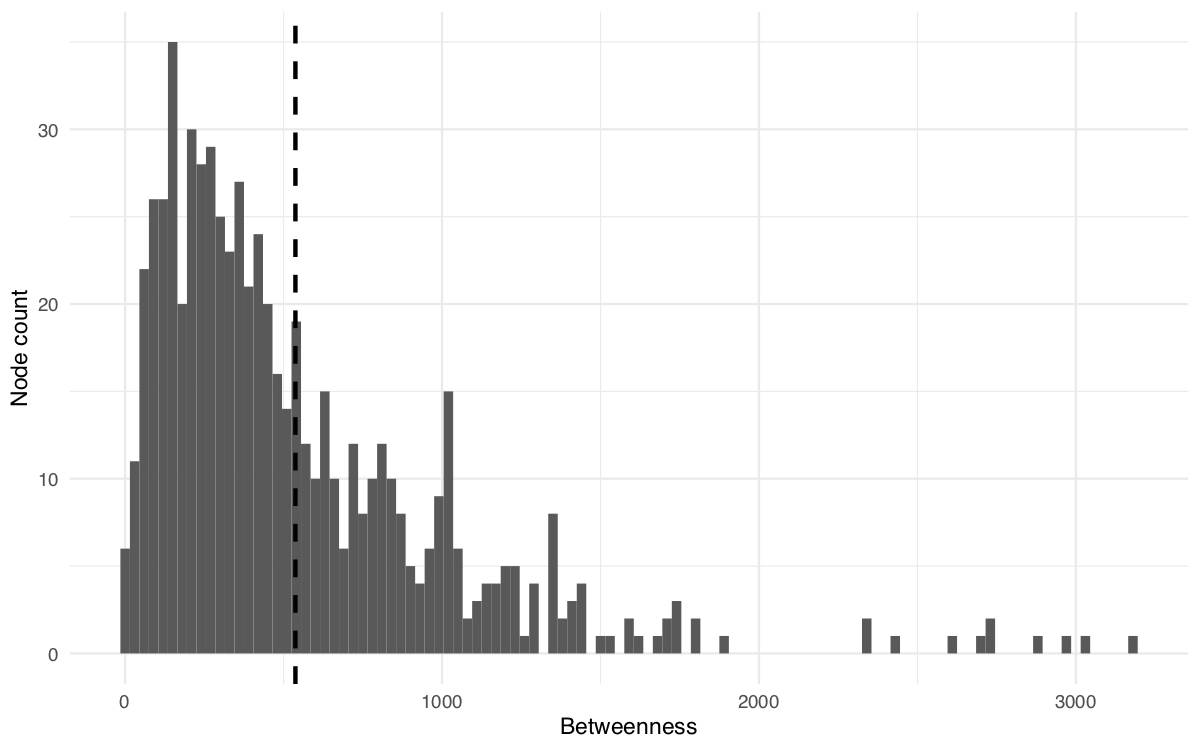
7.3 Keystone species
Como mencionamos antes, vamos a seleccionar como keystone species aquellos nodos que presenten los valores más altos en comparación a todos los nodos en la red.
- Comencemos por mirar ambos histogramas.
# Usamos la función 'grid.arrange' del paquete 'gridExtra' para visualizar ambos histogramas juntos
pdeg <- ggplot(deg_df, aes(x = deg_sort)) +
geom_histogram(binwidth = 1) +
scale_x_continuous(breaks=c(0,10,20,30,40,50,60)) +
geom_vline(aes(xintercept=mean(deg_sort)),
color="black", linetype="dashed", size=1) +
theme_minimal() +
labs(x = "Degree", y = "Node count", title = "Network Nodes Degree")
pbn <- ggplot(bn_df, aes(x = bn_sort)) +
geom_histogram(binwidth = 30) +
geom_vline(aes(xintercept=mean(bn_sort)),
color="black", linetype="dashed", size=1) +
theme_minimal() +
labs(x = "Betweenness", y = "Node count", title = "Network Nodes Centrality: Betweenness")
grid.arrange(pdeg, pbn, ncol = 2)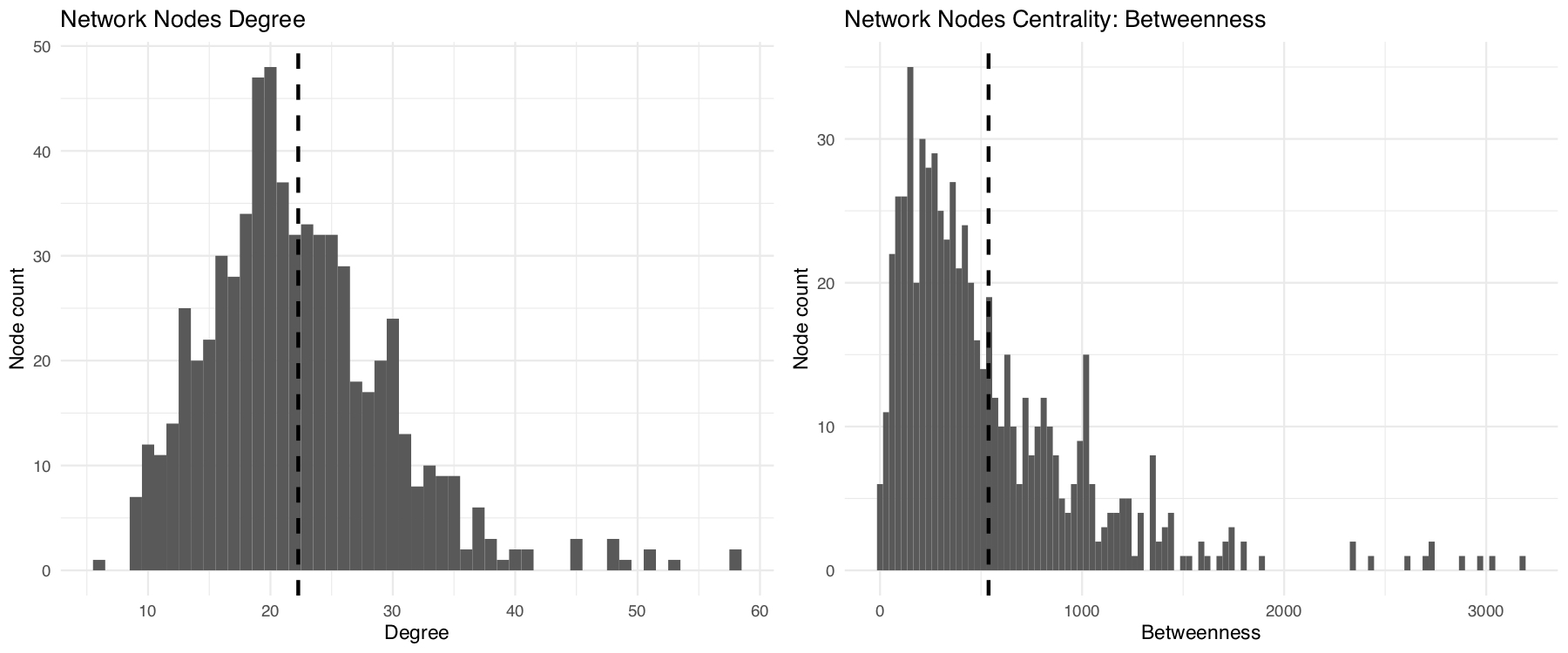
Basándonos en la distribución de los valores de degree y betweenness, vamos a definir como criterio:
Degree > 30
Betweenness > 1000
- Hacemos la selección siguiendo el criterio definido arriba.
# Editar la tabla de degree
head(deg_df)## deg_sort
## TI_27397 58
## TI_27345 58
## TI_27344 53
## TI_27357 51
## TI_27360 51
## TI_27772 49deg_df$TaxID <- row.names(deg_df)
head(deg_df)## deg_sort TaxID
## TI_27397 58 TI_27397
## TI_27345 58 TI_27345
## TI_27344 53 TI_27344
## TI_27357 51 TI_27357
## TI_27360 51 TI_27360
## TI_27772 49 TI_27772# Filtramos las taxas con degree > 30 y las guardamos en un nuevo data frame
deg_high_df <- dplyr::filter(deg_df, deg_sort > 30)
head(deg_high_df)## deg_sort TaxID
## 1 58 TI_27397
## 2 58 TI_27345
## 3 53 TI_27344
## 4 51 TI_27357
## 5 51 TI_27360
## 6 49 TI_27772# Editar la tabla de betweenness
head(bn_df)## bn_sort
## TI_27683 3184.922
## TI_27698 3026.017
## TI_38980 2980.506
## TI_29620 2889.191
## TI_23094 2724.146
## TI_27397 2723.078bn_df$TaxID <- row.names(bn_df)
# Filtramos las taxas con betweenness > 1000 y las guardamos en un nuevo data frame
bn_high_df <- dplyr::filter(bn_df, bn_sort > 1000)
head(bn_high_df)## bn_sort TaxID
## 1 3184.922 TI_27683
## 2 3026.017 TI_27698
## 3 2980.506 TI_38980
## 4 2889.191 TI_29620
## 5 2724.146 TI_23094
## 6 2723.078 TI_27397# Unimos las tablas de degree y betweenness filtradas
keystone <- merge(deg_high_df, bn_high_df, all.x = FALSE)
head(keystone)## TaxID deg_sort bn_sort
## 1 TI_12263 34 1719.243
## 2 TI_15981 38 1409.270
## 3 TI_19404 41 2595.900
## 4 TI_20829 31 1396.062
## 5 TI_22891 33 1214.696
## 6 TI_22895 34 1402.364# Ordenamos 'keystone' según degree de mayor a menor
keystone <- keystone[order(keystone$deg_sort, decreasing = TRUE),]
# Definir los ID de las taxas como row names del data frame 'keystone'
row.names(keystone) <- keystone$TaxID- El objeto
keystonecontiene los 53 nodos identificados como keystone species.
# Mira el objeto 'keystone'
View(keystone)| TaxID | deg_sort | bn_sort | |
|---|---|---|---|
| TI_27397 | TI_27397 | 58 | 2723.078 |
| TI_27772 | TI_27772 | 49 | 1304.233 |
| TI_27401 | TI_27401 | 48 | 1346.002 |
| TI_27698 | TI_27698 | 48 | 3026.017 |
| TI_27683 | TI_27683 | 45 | 3184.922 |
| TI_27771 | TI_27771 | 45 | 1891.286 |
| TI_19404 | TI_19404 | 41 | 2595.900 |
| TI_37462 | TI_37462 | 41 | 1669.543 |
| TI_27781 | TI_27781 | 40 | 1355.041 |
| TI_37729 | TI_37729 | 40 | 1445.233 |
- Ahora que ya tenemos nuestra selección de nodos representantes de keystone species en la red, vamos a visualizarlos en un gráfico de puntos según sus valores de degree y betweenness.
# Usamos ggplot2
ggplot(keystone, aes(x = bn_sort, y = deg_sort, label = TaxID)) +
scale_y_continuous(limits = c(30, 61), breaks = c(30,40,50,60)) +
scale_x_continuous(limits = c(1000, 3250), breaks = c(1000,1500,2000,2500,3000)) +
geom_point(alpha = 0.4, color = "#829FD9", size = 8) +
geom_text(size = 4) +
theme_minimal() +
labs(x = "Betweenness", y = "Degree",
title = "Nodes With Highest Degree And Betweenness: Keystone Nodes")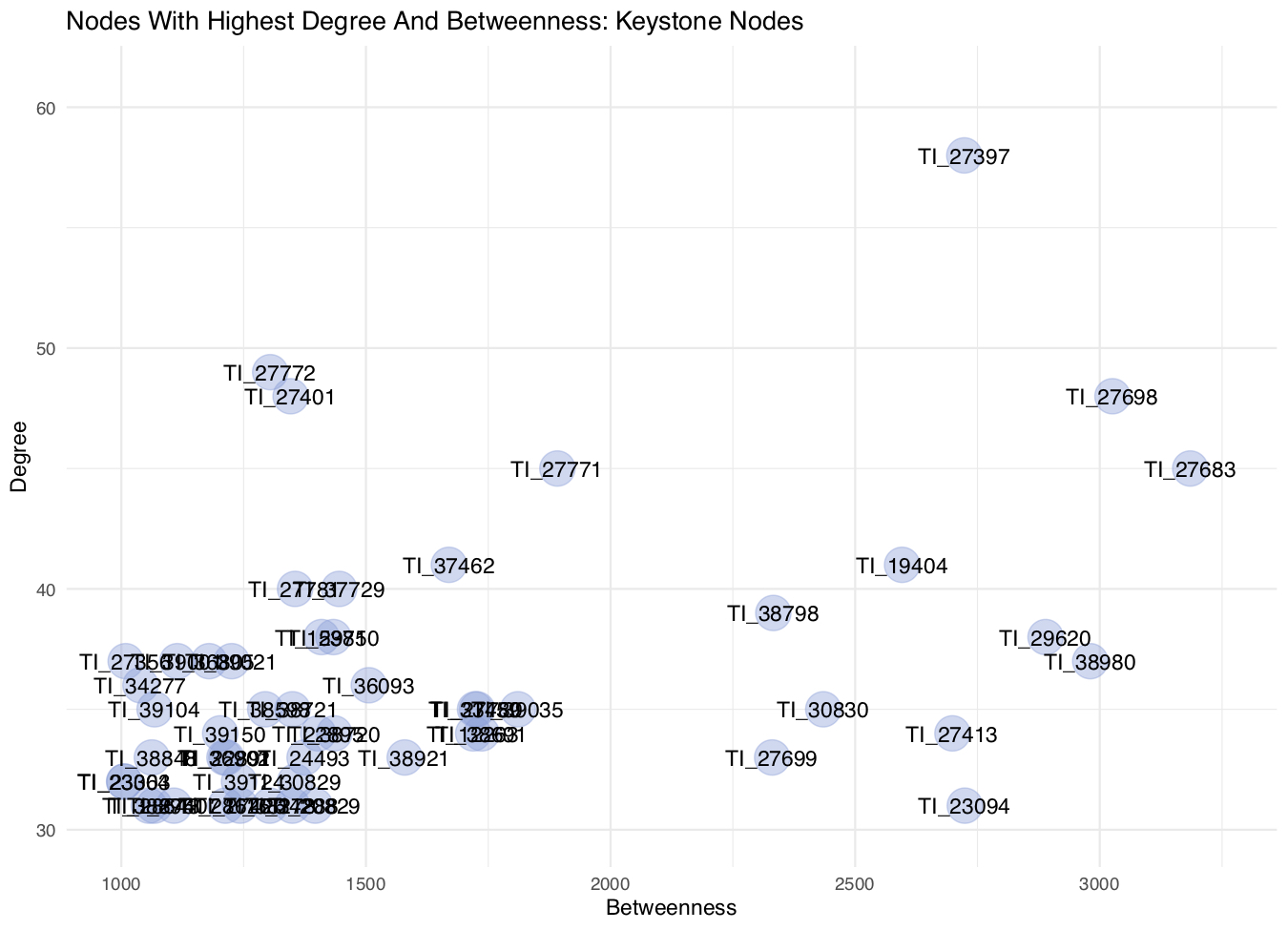
- Finalmente, vamos a visualizar la red de co-ocurrencia microbiana agregando una etiqueta (i.e., nombre del nodo/taxa) sobre los 53 nodos identificados como keystone species.
# Graficar red
ggnet2(net_class, mode = net$layout,
color = nodenames, palette = colors1,
node.alpha = 0.9,
edge.color = edge_colors,
label = keystone$TaxID, label.size = 4) +
stat_ellipse(aes(group = factor(wt$membership)), type = "norm")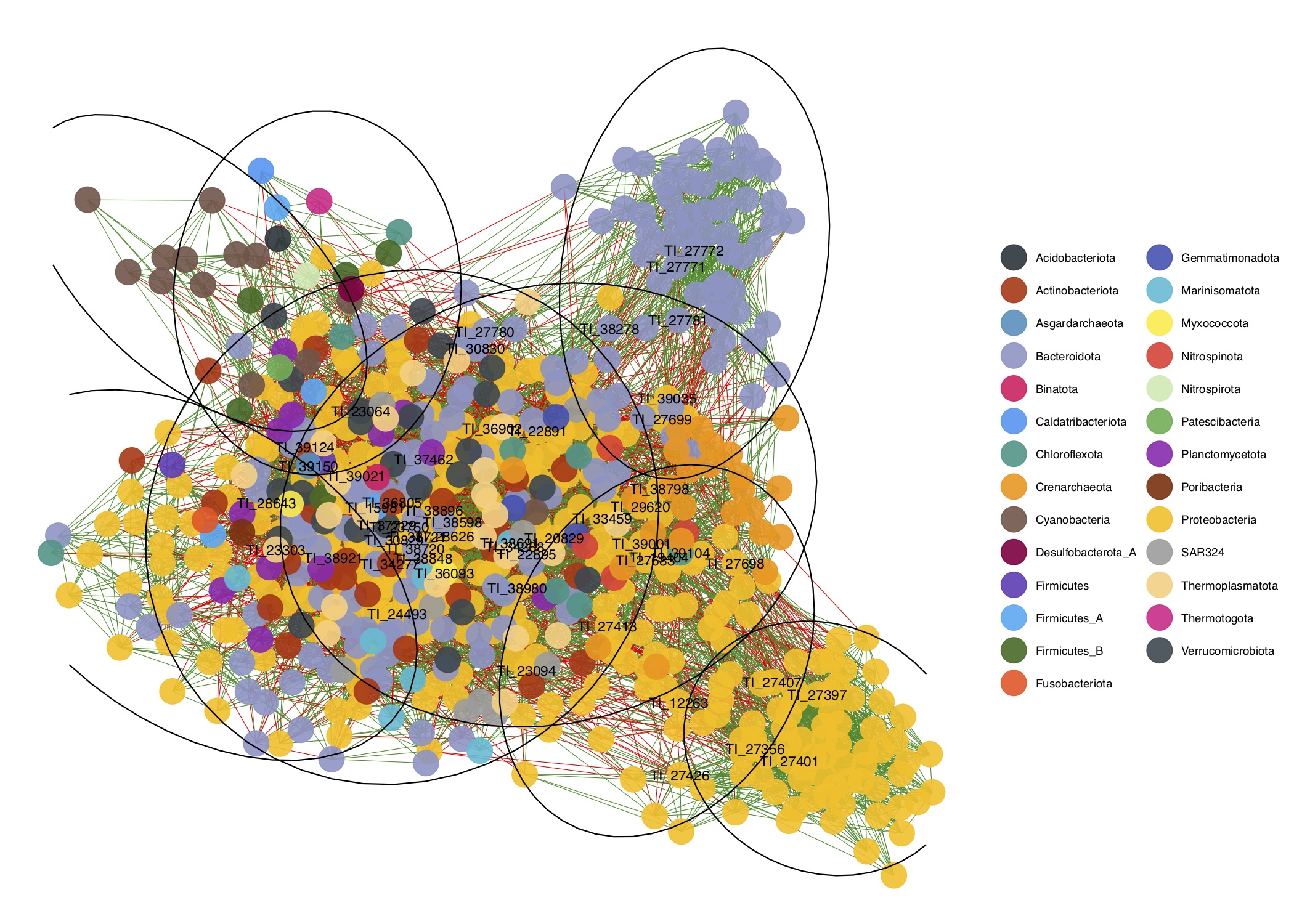
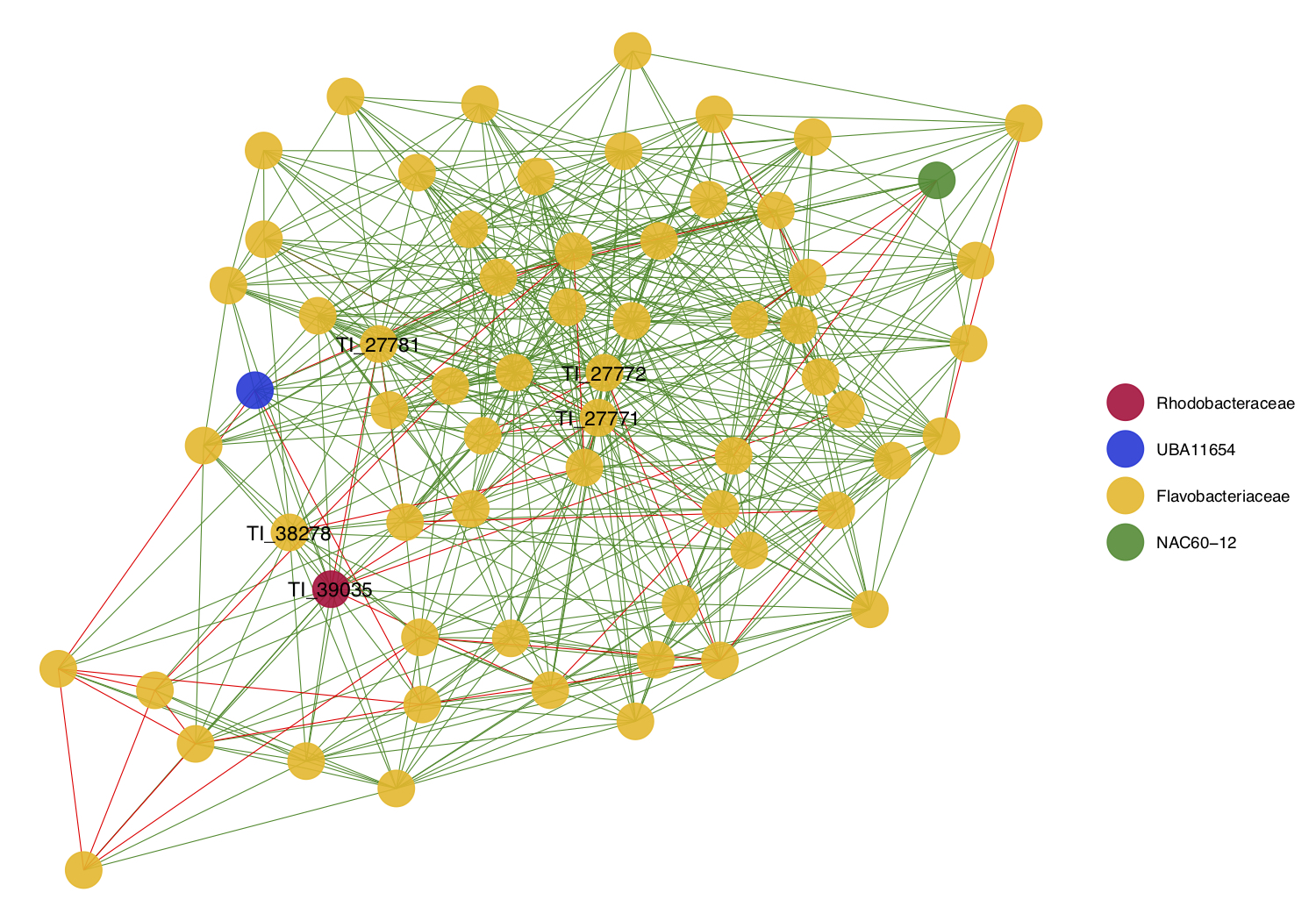
7.4 Nodo TI_27772
A continuación vamos a poner en práctica varias de las habilidades de análisis de redes que hemos aprendido en este tutorial para estudiar el nodo TI_27772.
Mira la última visualización de la red e identifica el nodo TI_27772, se trata de una keystone specie miembro de uno de los módulos de la red.
- Comencemos por identificar y extraer el módulo en el que se encuentra el nodo
TI_27772.
# Consultar de cuál módulo es miembro en nodo TI_27772
TI_27772 <- V(net)[wt$membership ==
wt$membership[which(wt$names == "TI_27772")]] # which position
TI_27772## + 64/650 vertices, named, from 1caee8a:
## [1] TI_39035 TI_34536 TI_27771 TI_27772 TI_27767 TI_27760 TI_27781
## [8] TI_27776 TI_27779 TI_27763 TI_27755 TI_27782 TI_27762 TI_27768
## [15] TI_27757 TI_27759 TI_27770 TI_27777 TI_27766 TI_27754 TI_27756
## [22] TI_27764 TI_27765 TI_27758 TI_27761 TI_27778 TI_27769 TI_27773
## [29] TI_33496 TI_33499 TI_33502 TI_33491 TI_33492 TI_33487 TI_33493
## [36] TI_33495 TI_33497 TI_33498 TI_33494 TI_33501 TI_33503 TI_33500
## [43] TI_33488 TI_38278 TI_38280 TI_22953 TI_22959 TI_22951 TI_22952
## [50] TI_22950 TI_22954 TI_22958 TI_22173 TI_22176 TI_33504 TI_13259
## [57] TI_13258 TI_18928 TI_36969 TI_18927 TI_13260 TI_31475 TI_13884
## [64] TI_13846# Inducir la sub-red correspondiente al módulo en el que se encuentra el nodo TI_27772
TI_27772_subnet <- induced_subgraph(net, TI_27772)
# Fijar el diseño de la sub-red 'TI_27772_subnet'
# Asignar coordenadas al diseño de la red
TI_27772_subnet$layout <- array(1:40, dim = c(20, 2))
# Asignar el diseño como un atributo fijo de la red
TI_27772_subnet$layout <- layout.fruchterman.reingold(TI_27772_subnet)
# Extraer matriz de adyacencia
TI_27772_class <- as_adjacency_matrix(TI_27772_subnet, type = "both")
# Generar objeto de clase 'network'
TI_27772_class <- network(as.matrix(TI_27772_class),
matrix.type = "adjacency", directed = F)- Graficamos la sub-red correspondiente al módulo del cuál el nodo
TI_27772es miembro.
ggnet2(TI_27772_class, mode = TI_27772_subnet$layout)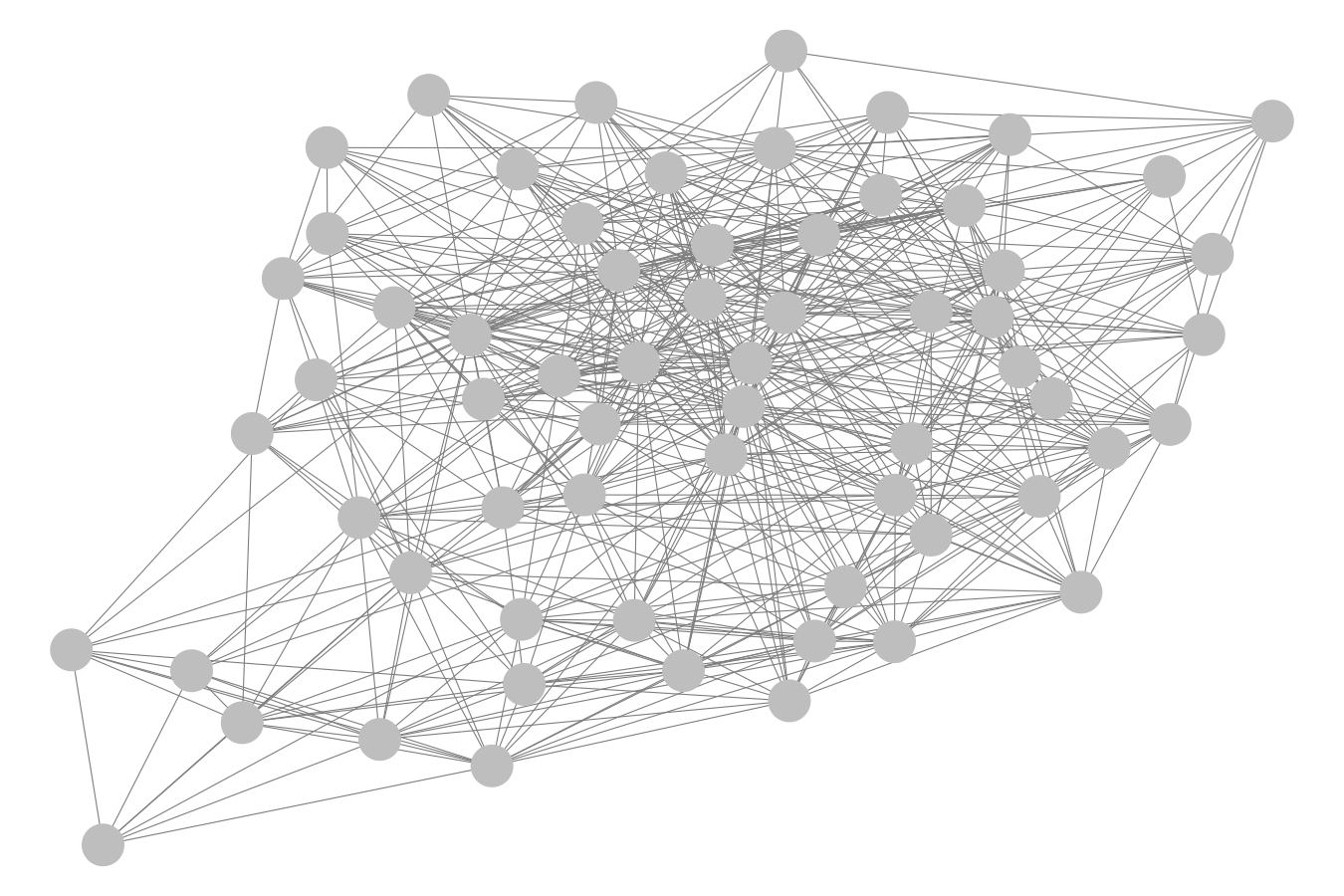
- Extraemos la clasificación taxonómica a nivel de familia de los nodos en la sub-red
TI_27772_subnet.
TI_27772_nodenames <- as.character(getTaxonomy(V(TI_27772_subnet)$name, tax_tbl, level = "family", useRownames = TRUE))- Graficar la sub-red
TI_27772_subnetcoloreando los nodos según su clasificación taxonómica a nivel de familia.
ggnet2(TI_27772_class, mode = TI_27772_subnet$layout, color = TI_27772_nodenames)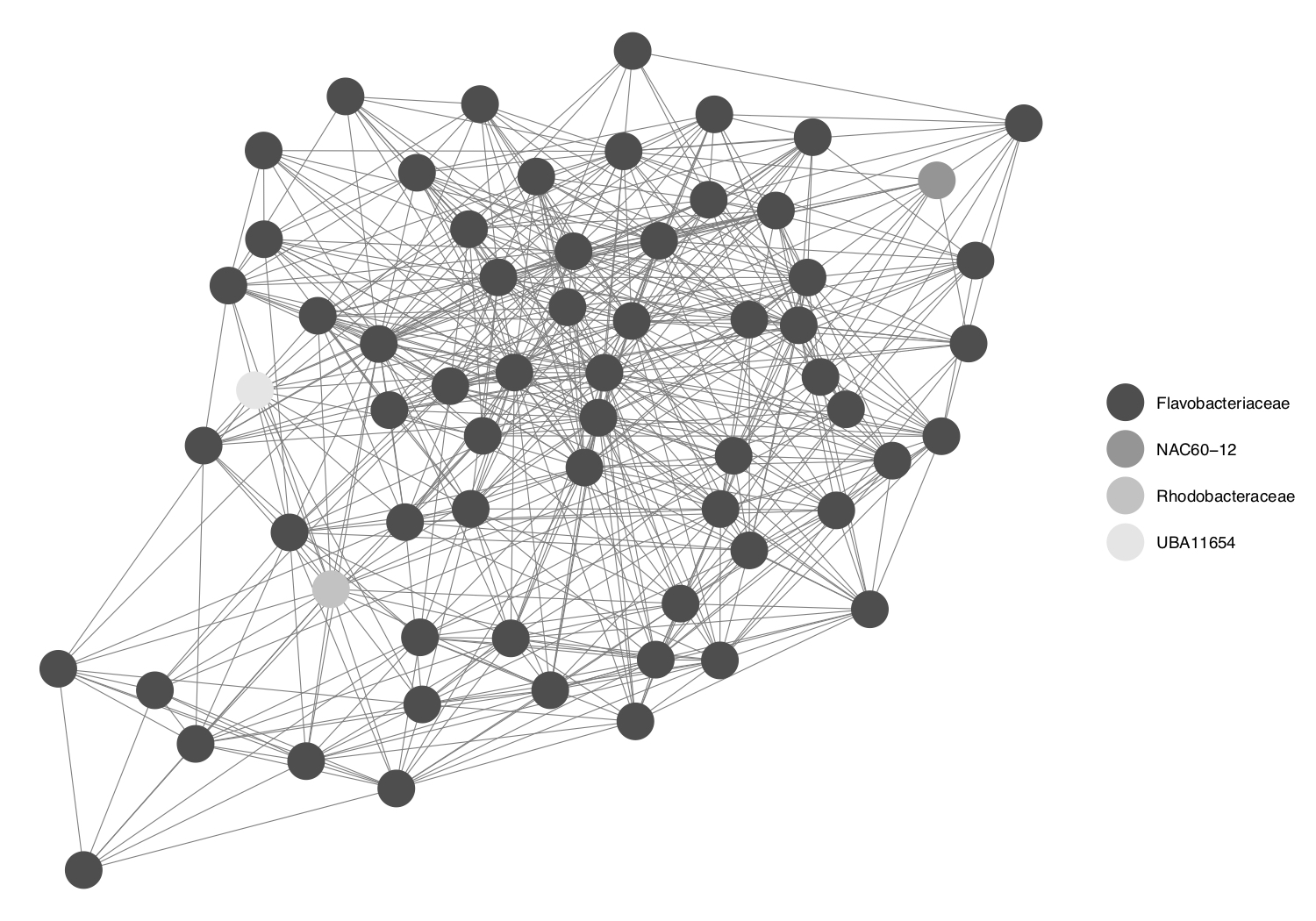
- Para agregar más información a la visualización de la sub-red
TI_27772_subnet; definimos una paleta personalizada de colores para colorear los nodos según su clasificación taxonómica a nivel de familia, y coloreamos los edges según si representan una relación positiva o negativa.
# Identificar familias presentes en la red
unique(TI_27772_nodenames)## [1] "Rhodobacteraceae" "UBA11654" "Flavobacteriaceae"
## [4] "NAC60-12"# Personalizar paleta de colores
colors2 <- c("Rhodobacteraceae" = "#BF0B3B",
"UBA11654" = "#1835D9",
"Flavobacteriaceae" = "#F2B90C",
"NAC60-12" = "#238C2A")
# Identificar y asignar color a los edges según si representan una relación positiva o negativa
edges1 <- E(TI_27772_subnet)
edge_colors <- c()
for(e_index in 1:length(edges1)){
adj_nodes <- ends(TI_27772_subnet,edges1[e_index])
xindex <- which(tax_ids==adj_nodes[1])
yindex <- which(tax_ids==adj_nodes[2])
beta <- betaMat[xindex,yindex]
if(beta>0){
edge_colors=append(edge_colors,"forestgreen") # positive
}else if(beta<0){
edge_colors=append(edge_colors,"red") # negative
}
}
E(TI_27772_subnet)$color <- edge_colors- Finalmente, visualizamos la sub-red correspondiente al módulo que contiene el keystone nodo
TI_27772.
ggnet2(TI_27772_class, mode = TI_27772_subnet$layout,
color = TI_27772_nodenames, palette = colors2,
node.alpha = 0.9,
edge.color = E(TI_27772_subnet)$color,
label = keystone$TaxID, label.size = 4)